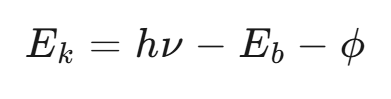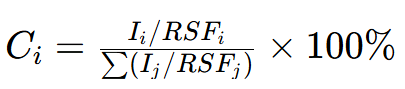说明:本文系统介绍 XPS 的定义、核心公式、典型应用场景、优势和局限性、分析方法以及它如何与其他表征技术(如XRD、HRTEM/STEM、EPR、LSV/EIS、Raman、XAS)相互印证,从而深入揭示材料结构–性能关系。
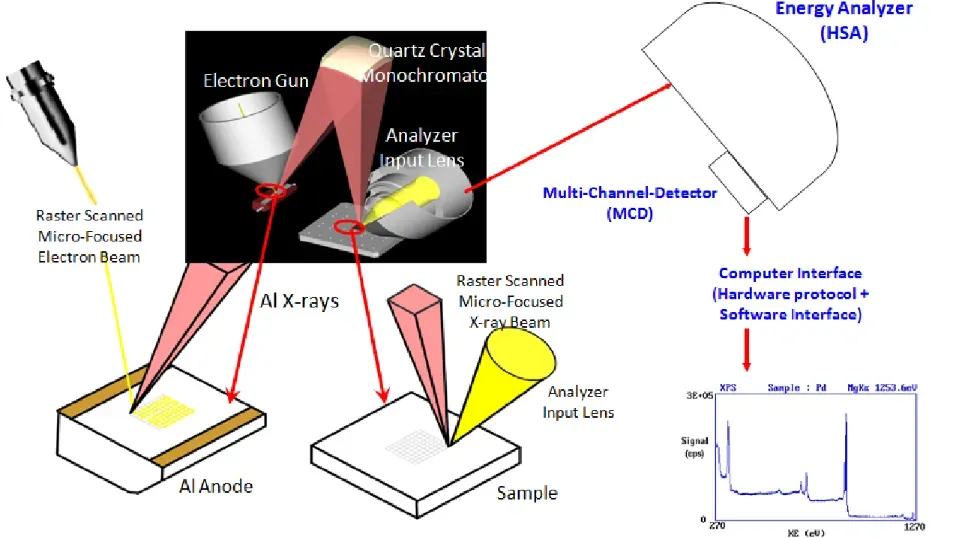
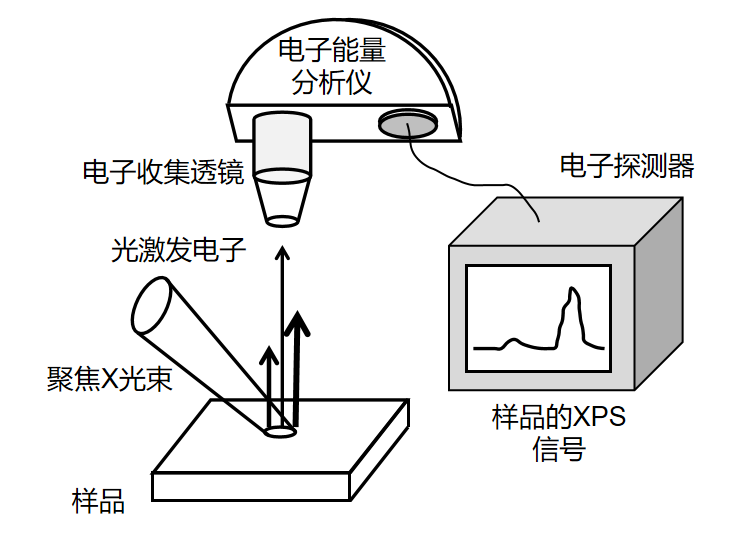
定义:X 射线光电子能谱(XPS)是基于光电效应的表面分析技术,通过照射样品表面的单色 X 射线激发光电子,测量光电子的动能与强度,推导样品表面元素的组成、化学价态及电子结构。
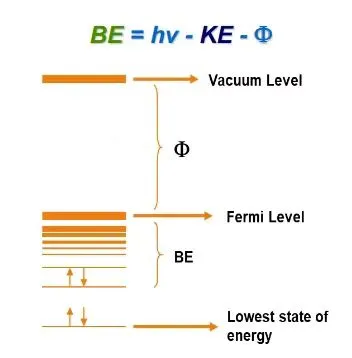
其中,Ek 为光电子动能,hν 为入射X射线能量,Eb 为结合能,ϕ 为仪器功函数。
典型应用场景:揭示催化材料的活性位点与电子协同机制;表征薄膜与涂层材料的界面组成与厚度均匀性;解析电子材料与器件的表面态与接触电阻机制;评估生物医药材料的生物相容性与表面功能化。
优势:分析维度全面,能够兼顾元素组成、价态与电子结构;表面选择性强;保护样品原始状态;空间分辨率灵活。
局限性:仅反映表层信息,无法表征体相;空间分辨率有限,难以表征纳米尺度局部区域;对样品导电性要求高;半定量为主,需依赖标准样品;无法分析挥发性或液态样品。
本文从 XPS 图谱的基础构成出发,详细解读全谱图、高分辨谱图及伴峰 / 卫星峰的核心信息。
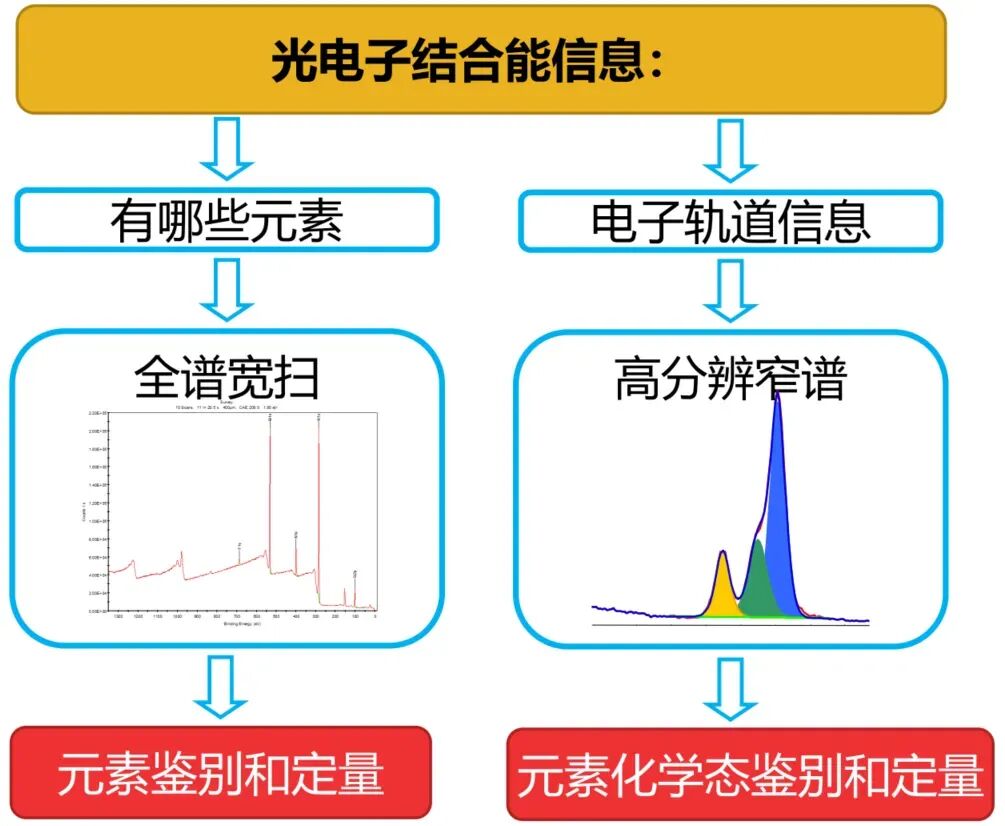
XPS 测试通常输出两类核心图谱:全谱图(Survey Scan) 与高分辨谱图(High-Resolution Scan) ,二者功能互补,分别对应 “元素定性与定量” 和 “价态与电子相互作用分析”。
全谱图是一张宽能量范围的扫描图谱,目的是定性分析样品表面存在的所有元素(除H和He外)。
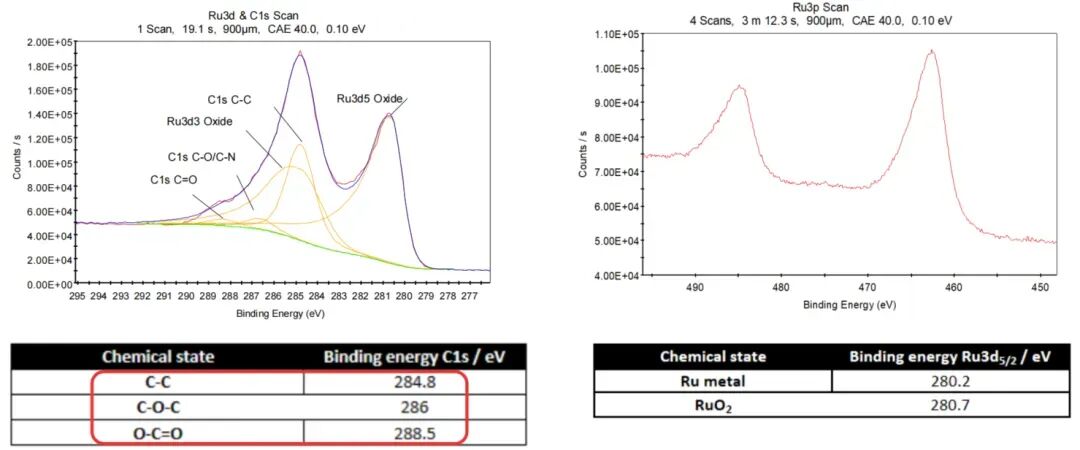
光电子峰:每个元素都有其特定的核心能级(如1s, 2p, 3d等),这些能级被激发会产生特征峰。在全谱中,你会看到代表不同元素的特征峰群。例如:Cu 2p, Cu 3p, Cu 3s 峰表明铜的存在。如果样品有污染,常会看到 C 1s 和 O 1s 峰。
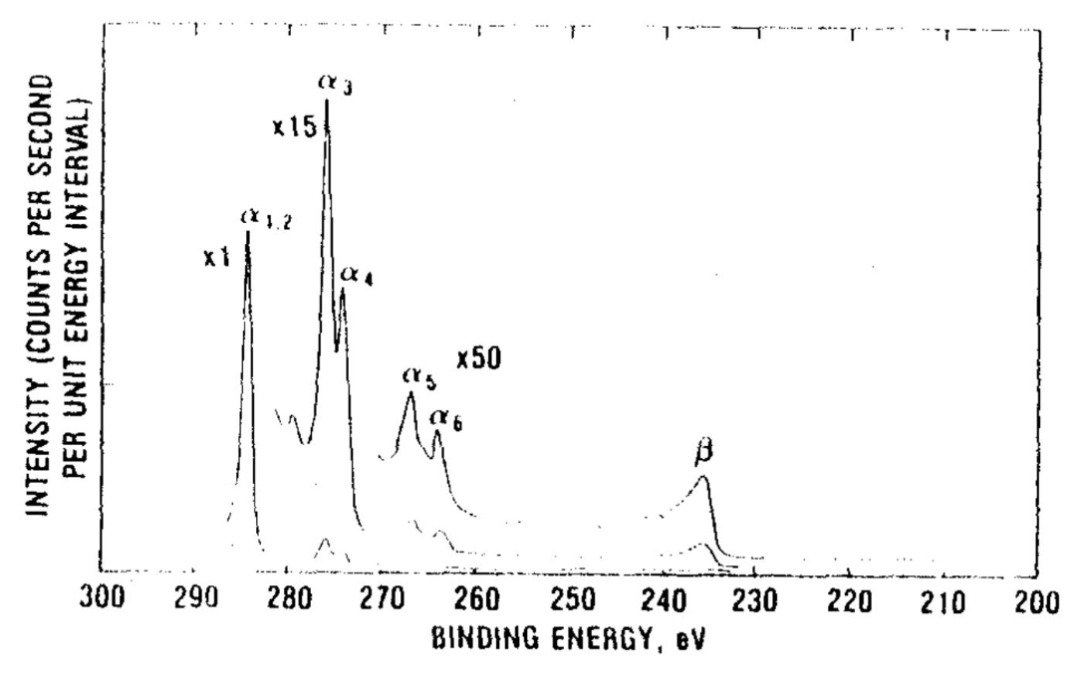
俄歇电子峰:元素还具有特征性的俄歇电子峰,通常用“元素字母+KLL/LMM”等标记,如 Cu LMM, O KLL。这些峰对化学态也非常敏感。
能量位置(横坐标):结合能(Binding Energy),单位是电子伏特(eV)。
信号强度(纵坐标):计数(Counts per Second),代表检测到的光电子数量,与元素含量成正比。
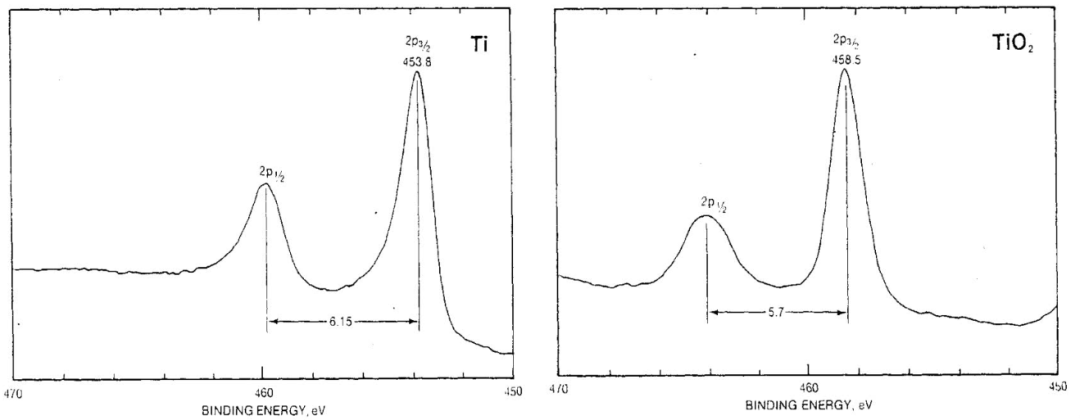
光电子峰:Ti及TiO2中2p3/2峰的峰位及2p1/2和2p3/2之间的距离。
高分辨率谱图是在确定元素组成后,对特定元素的特征峰进行精细扫描得到的图谱。目的是确定该元素的化学态和化学环境。
主峰:例如,对Cu元素,会精细扫描 Cu 2p 区域。峰的位置(结合能)、形状和卫星峰可以精确反映元素的化学价态和成键情况。
峰位(结合能):这是化学态分析的关键。结合能的位移(化学位移) 意味着元素化学环境的变化。
负移(结合能降低):通常表示元素周围电子密度增加(如被还原,或与吸电子基团分离)。
正移(结合能升高):通常表示元素周围电子密度降低(如被氧化,或与吸电子基团键合)。
峰形和卫星峰:对于某些元素(如Cu²⁺),会有明显的振离卫星峰,而Cu⁺或Cu⁰则没有,这是区分价态的重要依据。
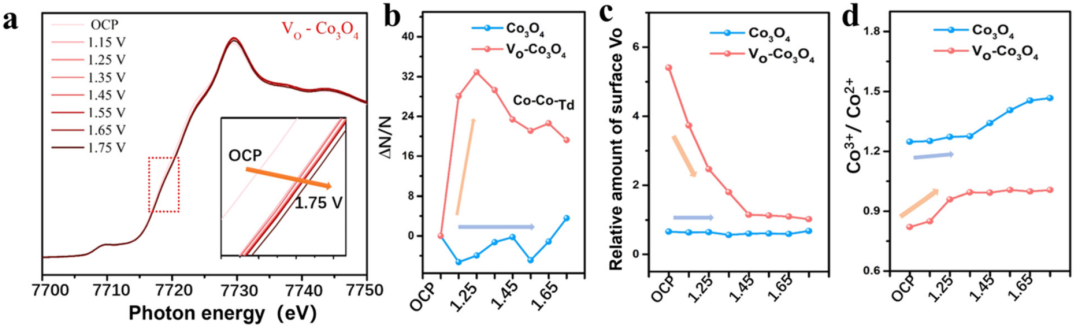
(d)VO-Co₃O₄的 Co 2p 高分辨谱图。DOI: 10.1039/d4cs00217b
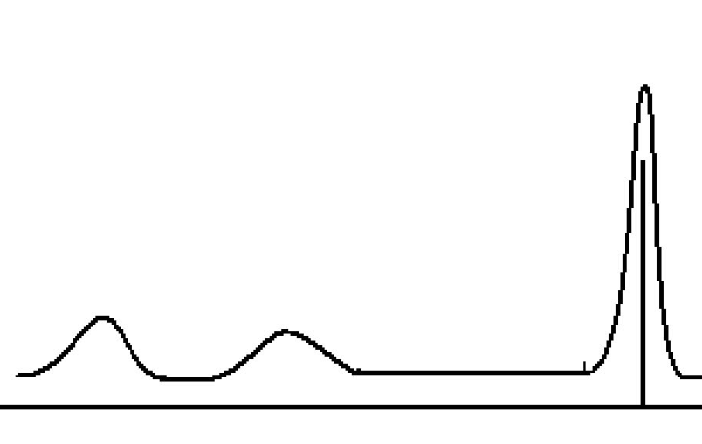
除主特征峰外,XPS 图谱中还存在伴峰(Shake-up Satellites) 、卫星峰(Satellite Peaks) 与背景信号。
伴峰(又称 “shake-up 峰”)源于光电子激发时,外层电子吸收能量跃迁至空轨道(shake-up 过程),导致光电子动能降低、结合能升高,通常出现在主特征峰高结合能侧 5-20 eV 处,强度为主峰的 5%-20%。
在 Co 2p₃/₂高结合能侧~790 eV 处出现明显伴峰。DOI: 10.1039/d4cs00217b
卫星峰(又称 “化学卫星峰”)与伴峰不同,主要源于晶格畸变或缺陷导致的电子能级分裂,常见于存在强电子相互作用的体系(如氧化物、硫化物中的缺陷区域)。
其特征为:峰位置靠近主特征峰,强度较弱;与缺陷密度正相关,缺陷越多(如氧空位、晶格畸变),卫星峰强度越强。
XPS 图谱的背景信号主要源于非弹性散射电子(光电子在样品中传播时与其他原子碰撞,损失能量后形成的连续背景)。
其特征为:背景强度随结合能升高而降低;主特征峰附近背景呈 “阶梯状”,需通过背景扣除才能准确计算峰面积。
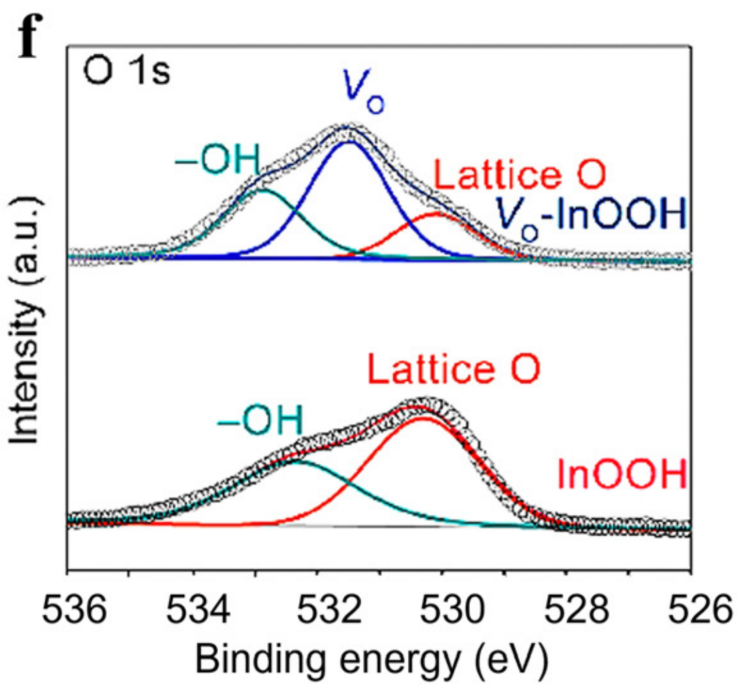
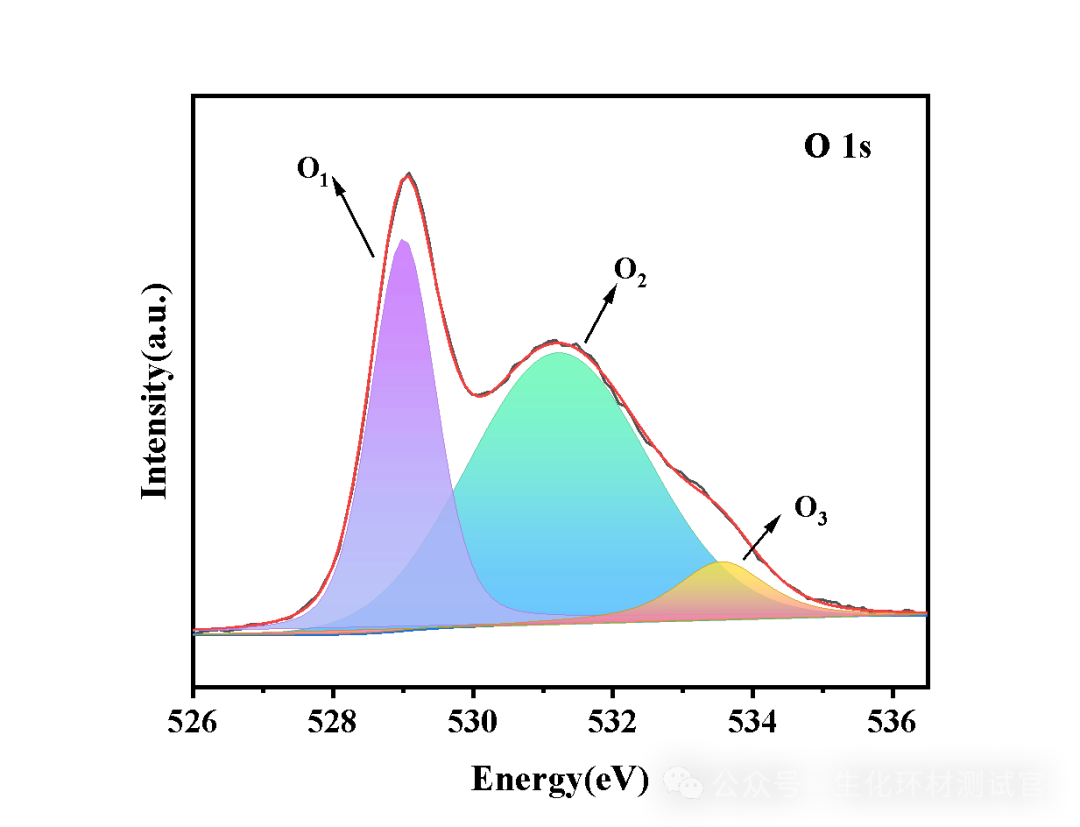
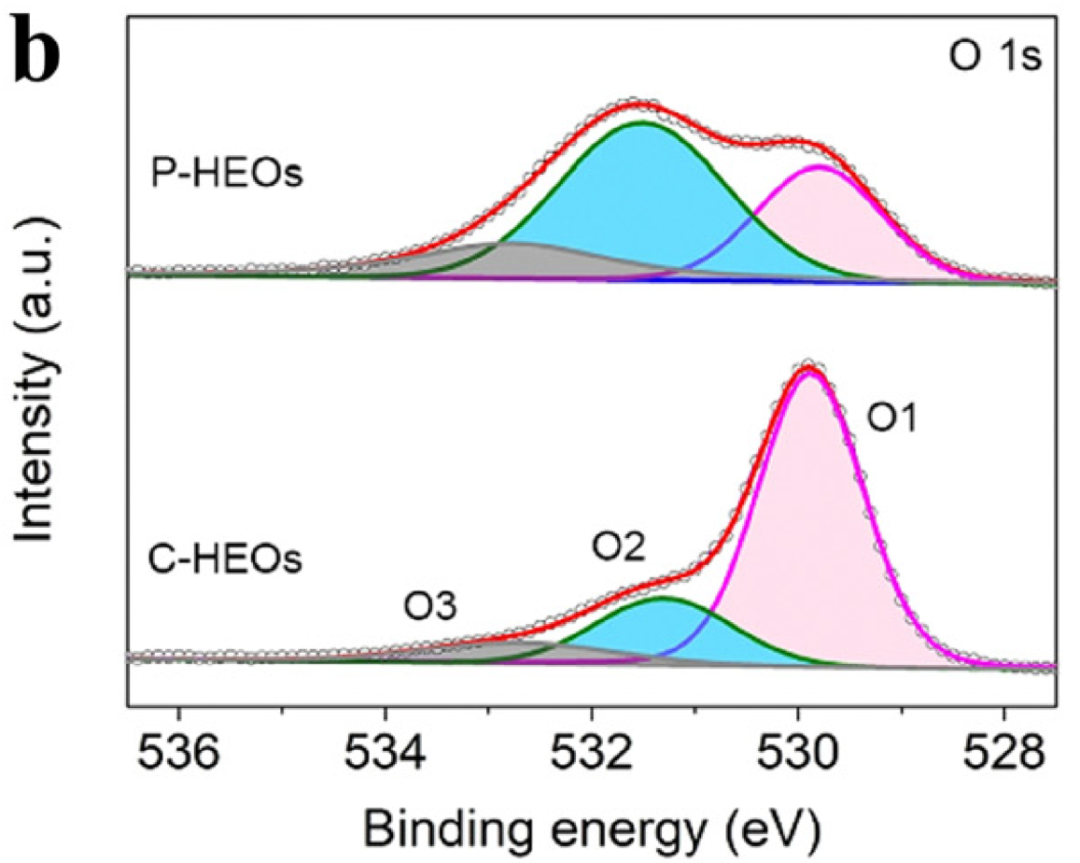
在 528 eV(背景起点)与 535 eV(背景终点)之间拟合 Shirley 背景,扣除后 O 1s 的晶格氧峰、缺陷氧峰与吸附氧峰清晰分离,确保空位浓度计算的准确性。DOI: 10.1039/d4cs00217b
当一个元素的峰是由多种化学环境的原子贡献时(例如处于不同价态,或与不同原子成键),这个峰可能是不对称的或明显展宽的。
此时需要用软件进行分峰拟合,将其分解为多个子峰,每个子峰代表一种特定的化学态。
拟合包络线(envelope):所有子峰叠加后形成的总曲线,应尽可能与原始数据重合。
子峰:每个子峰用峰位(P)、峰面积(A)、峰宽(W)和峰形(如高斯-洛伦兹混合比) 来定义。
峰面积:正比于该化学态原子的数量,可用于半定量分析。
XPS分析是一个系统性过程,需通过 “谱图预处理→定性分析→定量分析→价态与电子结构解析” 的逻辑链,将原始谱图数据转化为材料表面化学组成、价态及电子相互作用的科学结论。
在开展分析前,需明确样品特性与谱图类型,为后续分析奠定基础:
1.样品特性:确认样品导电性,非导电样品易产生 “荷电效应”,需提前规划校准方案;确认样品是否含杂质,若存在杂质,需在分析时排除杂质峰干扰。
2.谱图类型区分:全谱图输出 “元素有无” 与 “相对含量初步趋势”;高分辨谱图用于解析化学价态、电子相互作用及峰分裂细节。
预处理是 XPS 分析的基础,核心目标是修正仪器误差与样品干扰,关键步骤包括荷电校正、背景扣除与峰校准:
荷电效应是 XPS 分析的常见干扰,尤其对非导电样品,需通过内标校正法或外标校正法进行校正。
注意事项:校正后需确保同一元素的特征峰位置符合标准数据库(如 NIST XPS 数据库),避免过度校正导致价态误判。
XPS 谱图中的背景信号源于 “非弹性散射电子”(光电子在样品中传播时能量损失),需通过背景扣除提取纯净的光电子信号。
常用扣除方法包括Shirley 背景扣除法(适用于宽峰、弱峰)和线性背景扣除法(适用于尖锐峰、对称峰)。
操作要点:基线点需选择 “无其他峰干扰的平缓区域”,避免包含相邻峰的信号导致背景扣除过度或不足。
不同仪器的 X 射线源与探测器效率存在差异,需进行能量校准和灵敏度校准。
定性分析的核心是通过特征峰位置判断样品中含有的元素,明确 “有什么元素”,关键步骤如下:
特征峰识别:对照 XPS 元素结合能标准数据库(如 NIST、Thermo Scientific 数据库),根据全谱图中峰的位置确定元素种类。
注意 “重叠峰区分”:部分元素的特征峰可能重叠(如 N 1s 与 C 1s 的卫星峰、O 1s 与 Si 2p),需结合高分辨谱图进一步验证;
杂质峰排除:若出现未知峰,需排查是否为仪器污染、样品制备残留,排除后再确认是否为目标元素。
峰分裂与自旋 – 轨道耦合验证:过渡金属(如 Co、Fe、Ni)的 2p、3p 谱图存在 “自旋 – 轨道分裂”(同一壳层电子因自旋角动量不同分裂为两个峰),分裂峰的间距与强度比固定,可作为元素存在的辅助证据。
化学环境初步判断:同一元素的不同化学环境对应不同结合能,例如 O 1s 峰中,529-530 eV 对应晶格氧(M-O 键),531-532 eV 对应缺陷氧或羟基氧,可通过峰位置初步判断元素的化学状态。
定量分析基于 “特征峰面积与元素含量正相关” 的原理,计算样品表面各元素的原子百分比,关键步骤如下:
净峰面积提取:背景扣除后,通过仪器软件积分计算特征峰的 “净峰面积”(仅包含光电子信号的峰面积,排除背景);
积分范围设定:积分范围需覆盖整个特征峰(从峰的起始基线到结束基线),例如 C 1s 的积分范围可设为 280-292 eV,确保包含所有自旋 – 轨道分裂峰与伴峰(若有)。
不同元素的光电子产率不同(重元素光电子产率高于轻元素),需引入 “相对灵敏度因子(RSF)” 校正,公式为:
RSFi:元素 i 的相对灵敏度因子(仪器手册提供,如 C 1s 为 0.25,O 1s 为 0.71)。
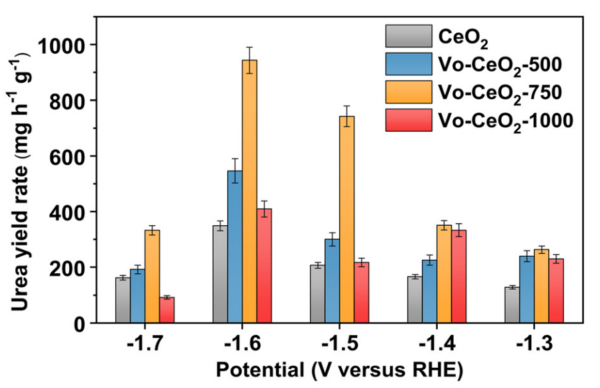
案例:计算VO-CeO₂的氧空位浓度,通过 O 1s 谱图分峰(晶格氧峰面积Alatt、缺陷氧峰面积Adef),定义空位浓度δ=Alatt+AdefAdef,结果显示VO-CeO₂的 δ 为 0.32,远高于原始 CeO₂的 0.05,解释了其优异的 C-N 耦合性能。
第四步:价态与电子结构解析 —— 揭示化学状态与相互作用
价态分析是 XPS 的核心,通过高分辨谱图的结合能偏移与峰分裂判断元素价态,关键在于:
特征峰位置对比:同一元素的不同价态对应不同结合能,对照已知价态元素的标准 XPS 谱图(如 NIST 数据库、文献报道),通过特征峰位置确定价态。
峰分裂与伴峰分析:部分元素的特征峰分裂与配位环境相关,通过分裂峰的间距与强度比可推导配位状态。
结合能偏移与电子相互作用推导:结合能偏移反映元素的电子云密度变化。结合能正移,代表电子云密度降低。偏移幅度越大,说明电子相互作用越强。
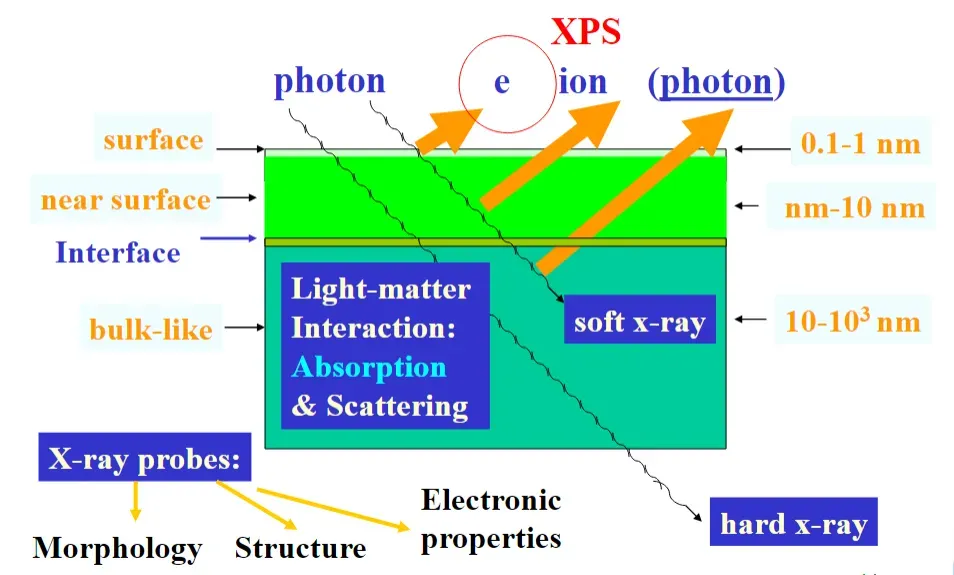
XPS虽能精准分析表面化学价态与电子相互作用,但存在分析深度浅、空间分辨率低等局限性。因此,“XPS + 多表征联动” 成为突破技术瓶颈、构建完整证据链的核心策略。
XPS+HRTEM/STEM:直接关联“化学态” 到 “形貌结构”
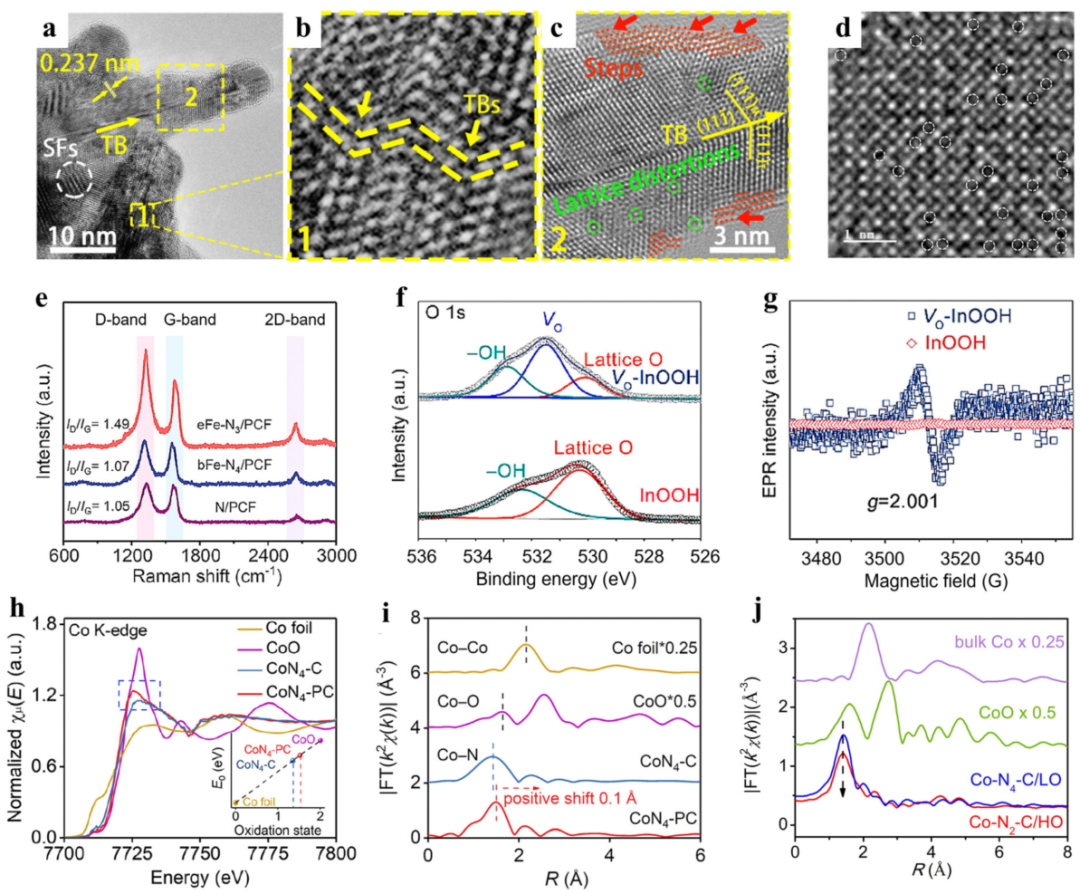
高分辨透射电子显微镜(HRTEM/STEM)可直接观察缺陷的形貌、尺寸与晶格结构(如氧空位导致的晶格畸变、掺杂原子的分散性),而 XPS 可解析缺陷区域的化学价态与元素分布。
二者结合可明确 “结构缺陷” 与 “化学状态” 的对应关系,避免仅靠形貌或化学态推断的片面性。
案例:HRTEM 观察到的晶格膨胀区域,对应 XPS 中缺陷氧峰强度最高的区域,证明氧空位的存在不仅导致晶格结构畸变,还改变了周围氧的化学环境,增强了 CO₂吸附能力(缺陷氧为 CO₂提供吸附位点),最终使 C-N 耦联生成尿素的产率达 943.6 mg h⁻¹ g⁻¹。
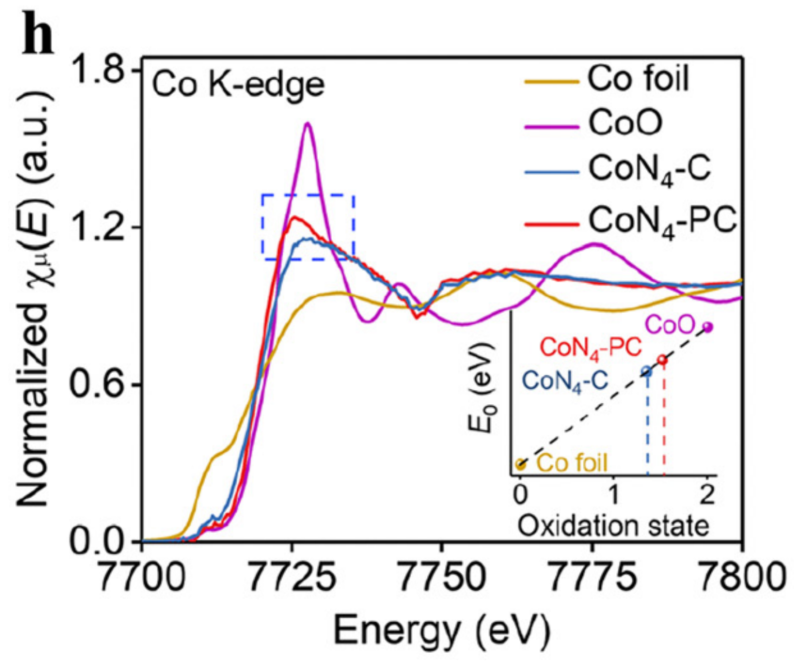
XPS+XAS:深度解析 “表面价态” 到 “体相配位”
X 射线吸收光谱(XAS)(含 X 射线吸收近边结构 XANES 与扩展 X 射线吸收精细结构 EXAFS)可揭示催化剂的体相电子结构与局部配位环境(如原子配位数、键长),而 XPS 仅反映表层(2-10 nm)的化学态。
二者结合可区分 “表面缺陷” 与 “体相缺陷”,明确缺陷对活性位点配位环境的调控机制。
案例:XPS 揭示的表层 Co 电子缺失,源于 XAS 观察到的 Co-N 键拉伸(键长增加导致电子云密度降低),这种配位环境优化使 * OOH 中间体的吸附能从 – 0.2 eV 降至 – 0.1 eV,提升了 H₂O₂电合成的选择性。
XPS+Raman:量化关联 “电子结构” 到 “缺陷密度”
拉曼光谱(Raman光谱)可通过特征峰的强度比量化缺陷密度(如碳材料的 D 带 / G 带强度比ID/IG,D 带对应缺陷,G 带对应石墨化),而 XPS 可解析缺陷区域的电子相互作用(如掺杂原子与基体的电荷转移)。
二者结合可建立 “缺陷密度 – 电子结构 – 催化活性” 的定量关系。
案例:Raman 的高ID/IG对应 XPS 中高比例的缺陷碳与吡啶 N,二者共同作用使石墨烯的功函数从 4.6 eV 降至 4.2 eV(给电子能力增强),ORR 起始电位从 0.85 V 升至 0.94 V,接近商业 Pt/C(0.95 V)。
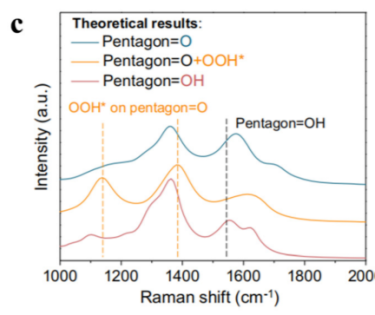
(c) 缺陷石墨烯表面三种可能的氧原子结构及相关物质的理论拉曼光谱。DOI: 10.1039/d4cs00217b
XPS+EPR:关联 “化学态” 到 “未成对电子” 的自旋状态
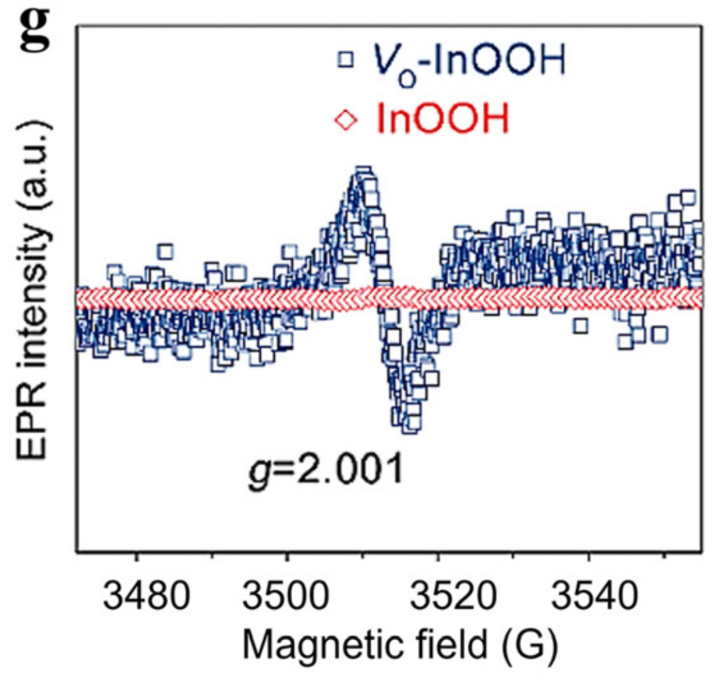
电子顺磁共振(EPR)可检测材料中的未成对电子(如氧空位中的孤对电子、过渡金属的未成对 d 电子),直接证明缺陷的存在;而 XPS 可解析缺陷区域的化学价态。
二者结合可明确 “未成对电子来源” 与 “化学态” 的对应关系,揭示缺陷的电子自旋对催化性能的影响。
案例:EPR 的未成对电子信号对应 XPS 中的氧空位缺陷峰,这些未成对电子可稳定*NO 中间体(尿素合成的关键中间体),避免其质子化,使尿素选择性从 35% 增至 72%。
XPS+原位LSV/EIS:关联 “静态化学态” 到 “动态催化过程”
原位线性扫描伏安法(LSV)、电化学阻抗谱(EIS)可实时监测催化剂的催化活性与电荷转移能力,而准原位 XPS 可捕捉反应不同阶段的表面化学态变化。
二者结合可揭示 “催化过程中缺陷的动态演化”,明确真正的活性物种。
案例:原位 EIS 的电阻降低与准原位 XPS 的 Ni²⁺→Ni³⁺、空位消耗一致,证明阳离子空位诱导 NiOOH 活性物种生成,这种 “动态重构” 使 OER 过电位从 310 mV 降至 270 mV。
本文源自微信公众号:材料有干货
原文标题:《如何分析XPS??》
原文链接:https://mp.weixin.qq.com/s/9nPX4QipYrx_wMwb9QpZYw
本转载仅出于分享优质测试干货,旨在传递更多观点,并不代表赞同其全部观点或证实其内容的真实性。文章中所包含的图片、音频、视频等素材的版权均归原作者所有。如有侵权请告知删除。