

VASP(Vienna Ab initio Simulation Package)是一种广泛应用于密度泛函理论(DFT)计算的高效计算工具,其能带结构计算是研究材料电子性质的重要方法之一。本文华算科技将详细介绍如何使用VASP进行能带结构计算,包括准备输入文件、设置参数、计算步骤以及结果分析等内容,并附带相关图片说明。
准备工作
输入文件准备
在进行能带结构计算之前,需要准备四个基本输入文件:INCAR、KPOINTS、POSCAR和POTCAR。这些文件分别用于控制计算参数、定义k点路径、描述晶体结构和指定赝势信息。
INCAR:包含计算控制参数,如电子迭代次数(IBRION)、自洽计算标志(ICHAR=11)、波函数截断参数(NBANDS)等。
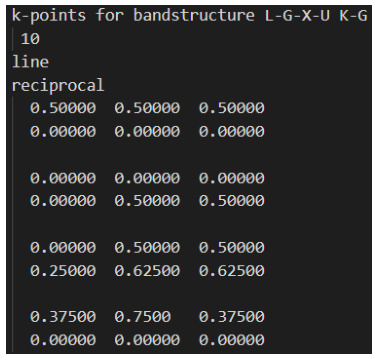
KPOINTS:定义k点路径,包括高对称性点(如G点、X点等)及其之间的分割点数,以决定能带的精细程度。
POSCAR:包含晶体结构的原子坐标和晶格常数,通常由结构优化后的结果生成。

POTCAR:包含赝势文件,用于描述原子的电子相互作用。
文件命名与目录管理
将上述文件复制到一个新的计算目录中,并根据需要重命名文件,例如将INCAR重命名为INCARbands,KPOINTS重命名为KPOINTSbands等。
设置INCAR文件
在INCAR文件中,需要特别注意以下参数:
ICHAR=11:指定非自洽计算模式,读取波函数文件。
NBANDS:设置能带数量,通常取值为电子态数的两倍以上。
ISMEAR:设置高斯分布参数,用于平滑能带结构曲线。
LWAVE:开启波函数输出,以便后续提取数据。
生成KPOINTS文件
KPOINTS文件是能带计算的核心,其定义了k点路径和密度。对于Si等材料,推荐沿高对称性方向(如G点到X点)设置多个k点,以提高计算精度。
计算步骤
静态自洽计算
静态自洽计算是能带计算的前提,目的是获得体系的基态电荷密度和波函数。具体步骤如下:
使用INCAR、POSCAR和POTCAR文件运行VASP,执行静态自洽计算。
计算完成后,会生成CHGCAR、WAVECAR和OUTCAR等文件,其中CHGCAR记录电荷密度,WAVECAR包含波函数信息。
非自洽计算
非自洽计算是能带结构计算的关键步骤,其目的是基于静态自洽计算的结果,提取能带数据。
l将静态自洽计算生成的CHGCAR和WAVECAR文件复制到新的计算目录中。
修改INCAR文件,设置IBRION=0、ICHAR=11、ISMEAR=0等参数,并增加NBANDS以确保覆盖所有电子态。
运行VASP进行非自洽计算,生成EIGENVAL文件,其中包含了每个k点的本征值信息。
结果分析
非自洽计算完成后,EIGENVAL文件中包含了所需的能带数据。接下来需要对数据进行处理和可视化:
使用脚本工具(如PBND.x或Python脚本)将EIGENVAL文件转换为bnd.dat和highk.dat文件。
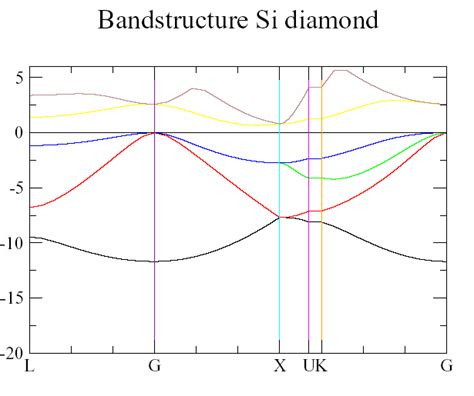
利用Origin或其他绘图软件读取bnd.dat文件,绘制能带结构图。
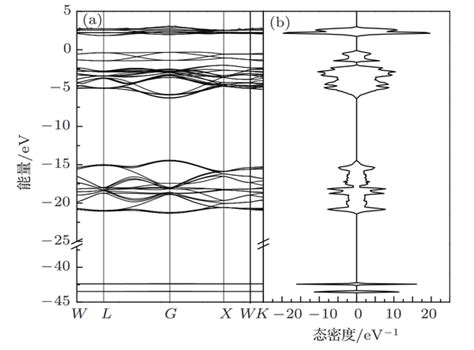
能带结构图绘制
数据提取
从EIGENVAL文件中提取每个k点对应的能量值,并根据POSCAR中的k点路径信息生成k路径坐标。
数据处理
使用PBND.x工具或Python脚本处理数据,生成bnd.dat和highk.dat文件。这些文件包含了标准化后的能带数据和k路径信息。
绘图
使用Origin或其他绘图工具加载bnd.dat文件,设置X轴为k路径坐标,Y轴为能量值(相对于费米能级),绘制能带结构图。
注意事项
k点路径选择
k点路径的选择直接影响能带结构的精度。建议沿高对称性方向设置足够多的k点,例如Si材料可设置G点到X点路径上的多个分割点。
NBANDS设置
NBANDS参数应足够大,以确保覆盖所有电子态。通常取值为电子态数的两倍以上。
电荷密度与波函数
静态自洽计算生成的CHGCAR和WAVECAR文件是能带计算的基础,需确保其准确性。
费米能级
能带结构图通常以费米能级为参考点(E=0),需从OUTCAR文件中提取费米能级值。
可视化工具
推荐使用Origin、Vasprun或Python脚本进行能带图绘制,这些工具支持多种数据格式并提供丰富的绘图选项。
总结
VASP能带结构计算是一个复杂但系统的过程,涉及输入文件准备、静态自洽计算、非自洽计算以及结果分析等多个步骤。通过合理设置参数、选择合适的k点路径以及准确处理数据,可以高效地获得高质量的能带结构图。本文结合多个实例和图片说明,详细介绍了VASP能带计算的全过程,希望能为相关研究提供参考。