

密度泛函理论(Density Functional Theory,DFT)是现代计算物理和材料科学中一种重要的理论方法,用于研究多电子体系的电子结构和性质。
在DFT计算中,自洽计算(Self-Consistent Field,SCF)是一个核心步骤,其目的是通过迭代求解Kohn-Sham方程,逐步逼近系统的基态电子密度和能量。本文华算科技将详细解释DFT中的自洽计算流程、原理及其应用,并结合多张图表展示其具体过程。
自洽计算的基本概念
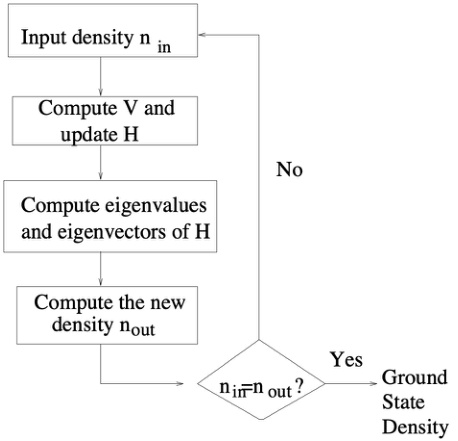
自洽计算是DFT计算的核心部分,其目标是通过迭代更新电子密度和有效势能,直到达到收敛状态。这一过程基于Kohn-Sham方程,该方程描述了在外部势场和交换关联势场作用下,单粒子波函数的行为。具体来说,自洽计算包括以下步骤:
初始化电子密度:从一个初始的电子密度分布开始。
计算有效势能:基于当前电子密度,计算系统的总势能,包括外部势能、哈特里势能和交换关联势能。
求解Kohn-Sham方程:根据有效势能,求解薛定谔方程以获得新的电子波函数。
更新电子密度:根据新的波函数计算电子密度。
判断收敛性:比较新旧电子密度的差异,若小于预设阈值,则认为达到自洽状态;否则返回步骤2继续迭代。


自洽计算的详细流程
自洽计算的流程可以通过以下步骤进行详细描述:
初始化电子密度
自洽计算从一个初始电子密度分布开始。这个初始密度可以是均匀分布、随机分布或基于经验的猜测。例如,在某些情况下,可以使用LDA(局域密度近似)或GGA(广义梯度近似)来初始化电子密度分布。
计算有效势能
根据当前电子密度,计算系统的总有效势能。有效势能由三部分组成:
外部势能:如电场或晶格势能。
哈特里势能:描述电子之间的静电相互作用。
交换关联势能:描述电子之间的交换和关联效应。

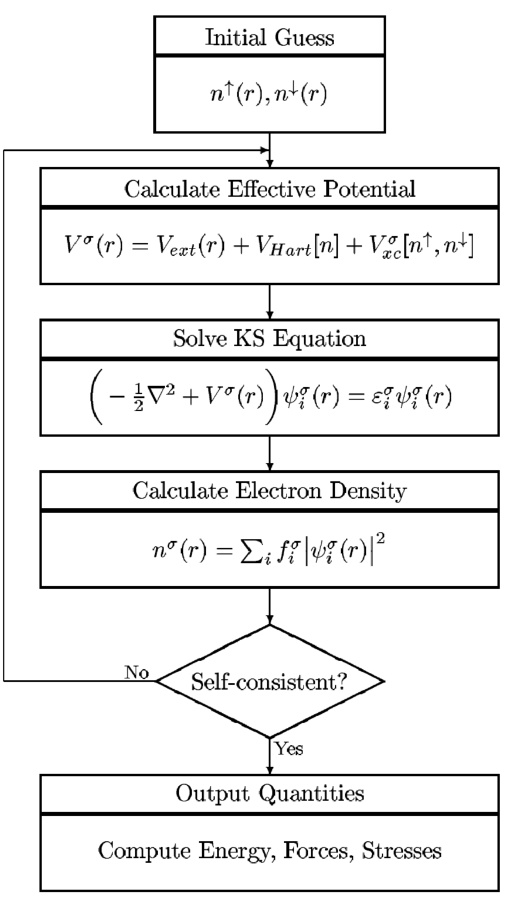
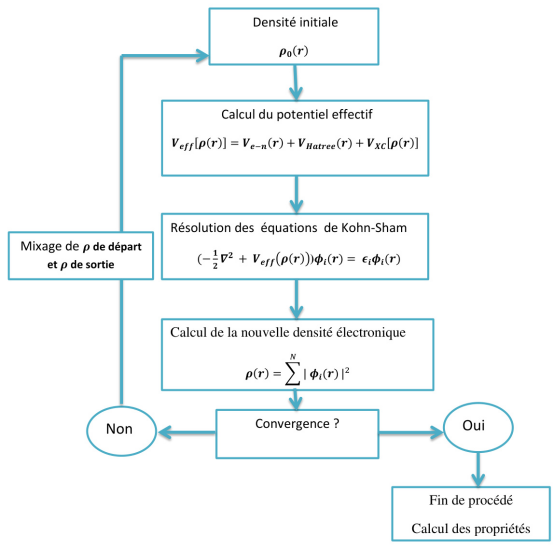
求解Kohn-Sham方程
使用有效势能,求解Kohn-Sham方程:
其中,是波函数,是对应的能量。通过求解该方程,可以得到一组基态波函数和对应的能量。
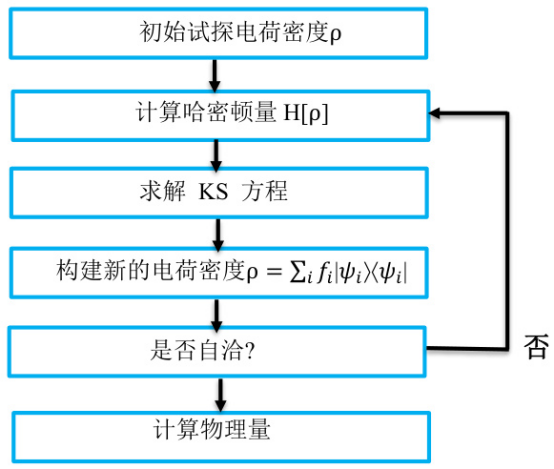
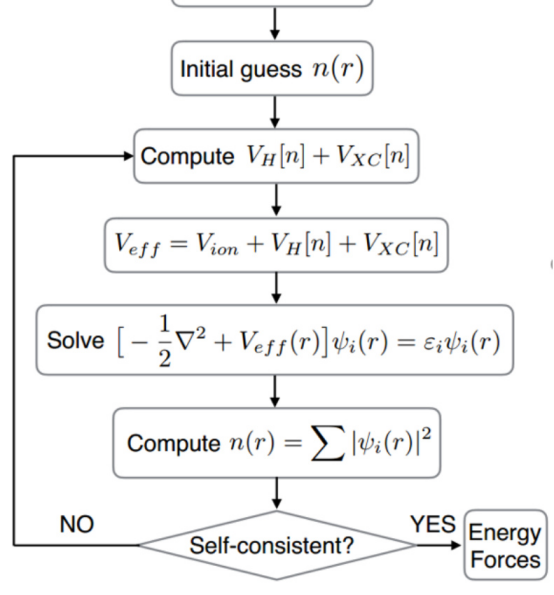
更新电子密度
根据求解出的波函数,计算新的电子密度:
其中,是占据因子(通常为1或0),表示电子是否占据对应的能级。
判断收敛性
比较新旧电子密度的差异,若满足收敛条件(如能量变化小于某个阈值),则认为计算达到自洽状态;否则返回步骤2继续迭代。
输出结果
当自洽计算完成后,输出最终的电子密度、能量、力和其他物理量。这些结果可用于进一步分析材料的电子结构、力学性质和化学反应活性。
自洽计算的应用
自洽计算在DFT中的应用非常广泛,包括但不限于以下几个方面:
材料设计与优化
自洽计算可用于预测材料的电子结构、力学性质和化学反应活性。例如,在催化剂研究中,通过模拟催化剂表面的吸附和反应过程,可以揭示反应机理并优化催化剂性能。
能带结构计算
自洽计算可用于计算晶体的能带结构和Berry相位等重要物理量。这些结果对于理解材料的导电性和磁性至关重要。
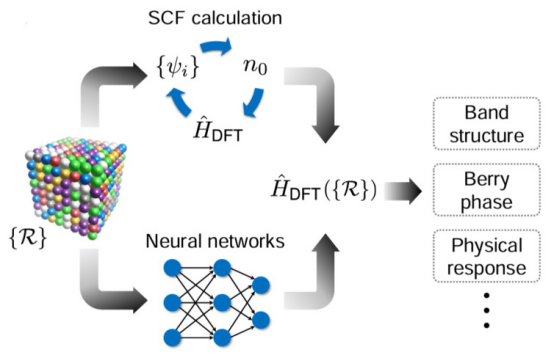
在分子动力学模拟中,自洽计算用于描述分子体系的电子结构和动力学行为。例如,通过模拟分子的振动模式和反应路径,可以研究化学反应的机理。
自洽计算在量子化学中用于研究分子的电子结构和反应性。例如,通过计算分子的HOMO(最高占据分子轨道)和LUMO(最低未占据分子轨道),可以预测分子的光学性质和反应活性。
多参考体系的研究
在强关联体系中,自洽计算可用于研究多参考体系的电子结构。例如,在强关联金属中,通过引入短程哈伯德型相互作用和残余项,可以更准确地描述电子间的交换-关联效应。
自洽计算的技术挑战与解决方案
尽管自洽计算在DFT中具有重要地位,但其计算过程也面临一些技术挑战:
收敛速度慢
对于某些复杂体系,自洽计算可能收敛缓慢。为解决这一问题,可以采用加速收敛技术,如引入Pulay混合器或预处理器。
数值精度问题
自洽计算对数值精度要求较高。例如,在处理大体系时,需要使用高精度的积分算法和大尺寸的基组。
系统性质的误判
在某些情况下,金属可能被误判为绝缘体。为解决这一问题,可以通过调整mixing_beta参数或选择更合适的mixing_mode来改善结果。
计算资源限制
对于大规模体系,自洽计算可能需要大量的计算资源。为解决这一问题,可以采用量子计算机或分布式计算平台。
总结
自洽计算是DFT计算的核心步骤,其目的是通过迭代求解Kohn-Sham方程,逐步逼近系统的基态电子密度和能量。自洽计算不仅在材料设计、能带结构计算和分子动力学模拟中具有重要应用,还在量子化学和多参考体系研究中发挥着关键作用。
尽管自洽计算面临一些技术挑战,但通过优化算法和提高计算资源利用率,可以有效克服这些问题。未来,随着计算技术的进步,自洽计算将在更多领域展现其潜力。