

VASP(Vienna Ab-initio Simulation Package)是一款广泛应用于材料科学领域的第一性原理计算软件,其在光学性质计算方面具有重要地位。本文华算科技将详细阐述如何使用VASP进行光学性质的计算,包括计算流程、所需文件、参数设置以及结果分析等内容,并结合相关文献和实例加以说明。
光学性质计算的基本原理
光学性质的计算通常基于介电函数的频率依赖性,介电函数可以描述材料对电磁波的响应。在第一性原理计算中,光学性质的计算主要通过以下步骤实现:
静态自洽计算:首先需要对材料进行结构优化和静态自洽计算,以获得准确的电子基态信息。这一步骤通常生成WAVECAR文件,这是后续光学性质计算的基础。
频率依赖介电函数的计算:通过广义布里渊区(GW)方法或独立粒子近似(RPA)方法,从电子结构数据中提取频率依赖的介电函数。这些方法能够考虑电子-电子相互作用的影响,从而更准确地预测材料的光学行为。
光学性质的提取与分析:通过计算得到的介电函数,进一步提取折射率、吸收系数、反射率等光学常数,并进行可视化分析。
VASP光学性质计算的具体步骤
准备输入文件
INCAR:设置计算参数,如算法选择(GW0或GW+BSE)、k点采样、电子态密度计算等。
POSCAR:包含优化后的晶体结构。
POTCAR:包含原子的伪势文件。
KPOINTS:定义k点路径,用于布里渊区采样。


运行静态自洽计算
使用VASP运行静态自洽计算,生成WAVECAR文件。这是后续光学性质计算的关键输入文件。
设置光学性质计算
在INCAR文件中设置LOPTICS=TRUE,启用光学性质计算功能。
设置NBANDS为电子态密度采样的带数,通常为默认值的两倍以上,以确保收敛性。
设置LWAVE=TRUE,用于更新WAVECAR文件中的频散量子态。
运行光学性质计算
运行VASP程序,生成包含光学信息的OUTCAR文件。
使用vaspkit工具读取OUTCAR文件中的介电函数数据,并生成REAL.IN和IMAG.IN文件。
结果分析与可视化
使用vaspkit生成折射率、吸收系数、反射率等光学常数文件(如REFRACTIVE.IN、ABSORB.IN等)。
l使用Origin或其他可视化工具绘制光学性质曲线,如吸收光谱和反射率曲线。
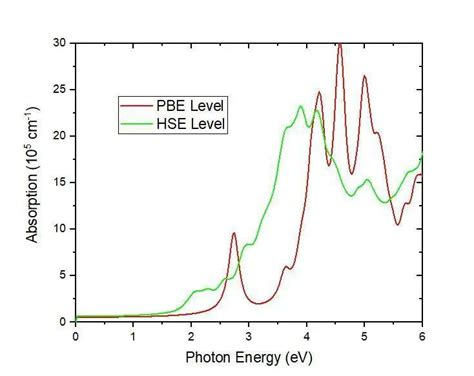
具体实例分析
以下以二维材料InSe为例,详细说明VASP光学性质计算的具体操作:
结构优化与静态自洽计算
使用VASP对InSe进行几何优化和静态自洽计算,生成优化后的POSCAR文件和WAVECAR文件。
设置INCAR参数,如ENCUT=400、ISMEAR=0、SIGMA=0.05等,确保计算精度。
光学性质计算
在INCAR文件中设置LOPTICS=TRUE,并调整NBANDS为216,以覆盖足够的电子态。
运行VASP程序后,使用vaspkit读取OUTCAR文件中的介电函数数据,并生成REAL.IN和IMAG.IN文件。
结果分析
使用vaspkit生成折射率和吸收系数文件,并用Origin绘制InSe的吸收光谱和反射率曲线。
分析结果表明,InSe在可见光范围内具有较高的吸收系数,表现出良好的光电性能。
注意事项与优化建议
k点采样密度
lk点采样密度直接影响介电函数的精度。建议使用高密度k点采样(如 Monkhorst-Pack 方法),以提高结果的准确性。
电子态密度采样
l设置合适的NBANDS值,通常为默认值的两倍以上,以确保电子态密度的完整性和收敛性。
算法选择
GW方法适用于半导体材料,而GW+BSE方法适用于金属材料。根据材料类型选择合适的算法。
计算时间与资源
光学性质计算通常耗时较长,建议使用高性能计算集群进行大规模计算。
总结
VASP作为一款强大的第一性原理计算工具,在光学性质计算方面具有广泛的应用前景。通过合理的参数设置和高效的计算流程,可以准确预测材料的光学性能。
本文结合具体实例,详细介绍了VASP光学性质计算的步骤和注意事项,并提供了相关文献支持。希望本文能为从事材料科学研究的学者提供有价值的参考。