

在计算催化自由能的过程中,VASP(Vienna Ab initio Simulation Package)是一种广泛使用的密度泛函理论(DFT)计算工具,它能够提供高精度的电子结构和能量计算。通过结合多种方法,如投影增强波(PAW)方法、广义梯度近似(GGA)以及分子动力学模拟,VASP可以有效地研究催化剂的结构、反应路径和自由能变化。



几何优化与结构弛豫
在计算催化自由能之前,首先需要对催化剂的几何结构进行优化。这一步骤的目的是确定催化剂在不同反应条件下的稳定构型。使用VASP进行几何优化时,通常采用以下步骤:
初始结构设置:根据实验数据或已有研究,构建催化剂的初始结构模型。例如,在研究M-N-C催化剂时,可以构建金属–氮–碳复合物的结构模型。
能量最小化:使用VASP计算不同结构的总能量,并通过几何优化方法(如BFGS算法)找到能量最低的结构。这一步骤确保了催化剂在计算中的稳定性。
结构弛豫:在优化过程中,VASP会自动调整原子的位置,使其达到能量最小值。这一过程通常需要多次迭代,直到能量变化小于设定的阈值。
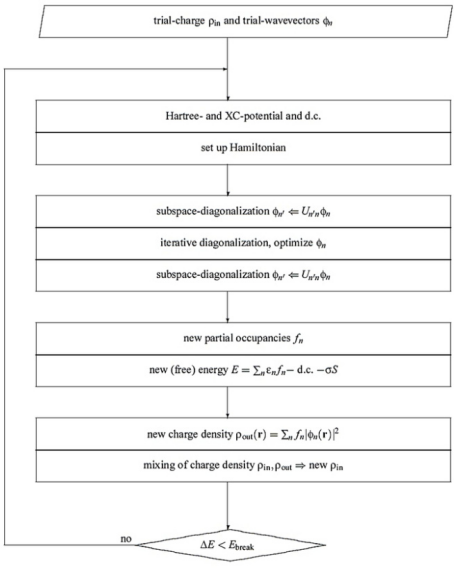
反应路径的计算
在确定催化剂的稳定结构后,下一步是计算反应路径,以确定反应的过渡态和活化能。VASP提供了多种方法来计算反应路径,其中最常用的是nudged elastic band (NEB) 方法。该方法通过在反应路径上引入一个“弹性绳”来寻找过渡态。
构建反应路径:在VASP中,可以使用nudge命令构建反应路径,并设置多个中间态。这些中间态代表了反应的不同阶段。
能量计算:VASP会计算每个中间态的总能量,并通过最小化能量来确定过渡态。这一过程通常需要多次迭代,直到能量变化小于设定的阈值。
过渡态验证:一旦找到过渡态,可以通过计算过渡态的频率来验证其稳定性。如果过渡态的频率为零或正频率,则说明该过渡态是稳定的。
自由能计算
在确定反应路径和过渡态之后,下一步是计算反应的自由能变化。自由能计算通常包括以下步骤:
总能量计算:使用VASP计算反应物、过渡态和产物的总能量。这些能量包括电子能量、离子能量和交换–关联能量。
热力学计算:根据热力学关系,计算反应的自由能变化。自由能变化可以通过以下公式计算:

其中,ΔH是焓变,ΔS是熵变,T是温度。
温度依赖性:在实际应用中,自由能通常需要在不同温度下进行计算。VASP可以使用phonopy软件计算振动频率,并通过Birch-Murnaghan方程拟合体积依赖的频率。此外,还可以通过热容重新缩放和积分来计算温度依赖的自由能。
动力学模拟与分子动力学
为了进一步研究催化剂的动态行为,可以使用VASP结合分子动力学(MD)模拟。分子动力学模拟可以提供催化剂在不同温度下的动态行为,从而帮助理解反应的微观机制。
初始配置:在分子动力学模拟中,首先需要构建催化剂的初始配置。这一步骤可以通过VASP计算得到的稳定结构进行设置。
动力学模拟:使用VASP的phonopy软件进行动力学模拟,计算催化剂的振动频率和热导率。这些数据可以用于计算热力学性质,如自由能和热容。
后处理:通过后处理软件(如ASE或VMD)对模拟结果进行分析,提取关键信息,如反应路径、过渡态和自由能变化。
机器学习与相图预测
在某些情况下,为了提高计算效率,可以使用机器学习方法来预测催化剂的相图和自由能。这种方法结合了VASP的高精度计算和机器学习的高效性。
训练数据生成:使用VASP计算不同催化剂的基态能量和振动频率,作为机器学习模型的训练数据。
模型训练:使用机器学习算法(如SchNetPack)训练模型,预测不同催化剂的自由能和相图。
相图预测:通过机器学习模型预测催化剂在不同温度和压力下的相图,并与实验数据进行对比。
总结
通过上述步骤,可以系统地计算催化剂的自由能,并结合实验数据进行验证。VASP作为一种强大的计算工具,能够提供高精度的电子结构和能量计算,为催化研究提供了重要的理论支持。未来,随着计算方法的不断进步,VASP在催化研究中的应用将更加广泛和深入。