




电荷转移量化:通过对比复合体系与孤立组分的电荷分布,直接反映原子间成键过程中电子的重新排布,例如离子键中的电子完全转移或共价键中的电子云重叠。
电子相互作用可视化:将抽象的量子化学相互作用转化为实空间的电荷密度差异分布,使化学键的极性、离域程度等特征可通过直观的图像呈现。
以半导体异质结为例,当分析 GaAs/AlAs 界面的差分电荷密度时,可观察到 Al 原子附近的电荷密度降低(电子转移)与 As 原子附近的电荷密度升高(电子聚集),这种分布直接对应 Al-Ga 原子间的电负性差异导致的电荷再分配,为界面偶极矩的计算提供了微观依据。


(一)第一性原理计算的基础设置
在密度泛函理论(DFT)框架下获取差分电荷密度需遵循严格的计算流程:
1. 模型构建与基组选择
周期性体系(如晶体)需建立超胞模型,非周期性体系(如分子吸附)需设置足够大的真空层(通常 > 15 Å)以避免周期性图像干扰。
基组选择直接影响电荷密度精度:平面波基组(如 PAW、 ultrasoft pseudopotential)适用于周期性体系,而 Gaussian 型基组(6-31G*、def2-TZVP)更适合分子体系。以 TiO₂表面 CO 吸附为例,采用 PBE 泛函 + PAW 势时,需将平面波截断能设为 400 eV 以确保电荷密度收敛。
2. 自洽场(SCF)计算收敛控制
能量收敛阈值通常设为 10⁻⁵~10⁻⁶ eV/atom,电荷密度收敛需满足总电子数变化小于 10⁻⁴ e。对于金属体系,需引入 Smearing 技术(如 Methfessel-Paxton,宽度 0.05 eV)以加速收敛。
(二)差分电荷密度的生成与后处理
1. 电荷密度文件的提取与处理
从 VASP、Quantum ESPRESSO 等软件输出的 CHGCAR、rho 文件中提取三维电荷密度数据,需注意不同软件的网格格式差异(如 VASP 的笛卡尔网格与 CP2K 的分数坐标网格)。
采用 Python 库(如 PyProcar、VaspTools)或可视化软件(VESTA、XCrySDen)对原始数据进行插值处理,将低分辨率网格(如 20×20×20)提升至适合可视化的精度(如 200×200×200)。
2. 差分运算的数学实现
空间网格点匹配是关键:需确保体系总电荷密度与各组分电荷密度的网格坐标完全一致,可通过线性插值或傅里叶变换实现不同网格间的数据映射。
对于包含多个原子的体系(如合金 Fe₃Al),需按原子类型分别提取各元素的孤立电荷密度,再通过原子位置映射到体系网格中进行叠加,避免坐标错位导致的计算误差。
(一)等值面与切片图的分析策略
1. 等值面图的定量分析
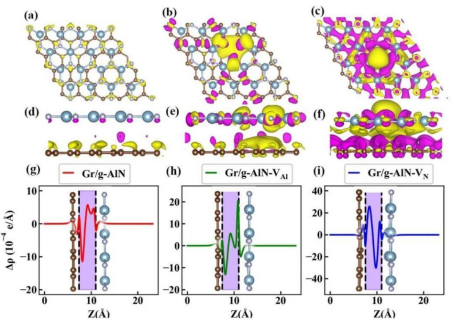
正电荷密度区域(Δρ>0)表示电子聚集,负电荷密度区域(Δρ)表示电子缺失,等值面阈值通常设为 ±0.01 e/ų 以平衡细节与噪声。在石墨烯 / 过渡金属界面体系中,正等值面可清晰显示金属 d 轨道与碳 p 轨道的杂化区域,而负等值面则指示金属原子的电子转移方向。
等值面的拓扑结构分析:通过计算等值面的连通性与曲率,可判断化学键的类型—— 如球状等值面对应离子键,哑铃状对应共价键,离域的片状结构对应金属键或 π 键。
2. 二维切片图的空间分布解析
选择特定晶面(如半导体的 (110) 面、分子吸附的表面法线方向)进行切片,可量化分析电荷密度的层间分布。以 MoS₂/WS₂异质结为例,沿层间垂直方向的切片图可显示范德华间隙内的电荷密度衰减速率,进而计算层间耦合强度。
结合颜色映射(如蓝–白–红表示 Δρ 从负到正),可直观识别电荷聚集的热点区域,例如在 CO 吸附于 Cu (111) 表面时,切片图中 C-O 键间的红色区域与 Cu 原子附近的蓝色区域形成鲜明对比,印证了 CO 的孤对电子向 Cu 的 d 带空穴转移的过程。
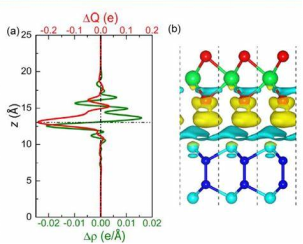
(二)电荷密度差分的定量表征方法
1. 积分电荷分析(Bader Charge)
通过 Bader 电荷划分方法,将差分电荷密度在原子分波函数区域内积分,可得到各原子的电荷得失量。例如在 Li 嵌入石墨体系中,Bader 分析显示每个 Li 原子失去 0.89 e,而相邻 C 原子获得 0.12 e,定量验证了 Li⁺与 C 的离子键特征。
注意事项:Bader 划分对网格精度敏感,需将电荷密度网格间距设为 以保证积分精度,同时需考虑原子间重叠区域的电荷归属算法(如 Wigner-Seitz cell 与 gradient-based 划分的差异)。
2. 电荷转移路径分析(CT Path)
利用 VESTA 的矢量场功能,将差分电荷密度与电子局域函数(ELF)结合,可追踪电子从供体到受体的转移路径。在有机太阳能电池给体 – 受体界面,这种分析可揭示激子解离过程中电子从 P3HT 的 HOMO 轨道向 PCBM 的 LUMO 轨道的迁移通道,为界面工程优化提供方向。
1. 电荷密度特征的自动化提取
利用卷积神经网络(CNN)对差分电荷密度图像进行特征识别,可自动分类化学键类型(如共价键、离子键、金属键)。训练集需包含上万组已知化学键类型的电荷密度图像,通过 ResNet 等模型学习等值面的几何特征(如伸长率、对称性),准确率可达 92% 以上。
2. 高通量筛选中的电荷密度指纹
在材料基因工程中,将差分电荷密度的统计特征(如总电荷转移量、电子聚集区域体积)作为描述符,结合随机森林算法构建预测模型,可快速筛选具有高催化活性的表面合金。例如在 OER 催化剂筛选中,电荷密度描述符可关联金属 – 氧键的离子性程度与反应能垒,将筛选效率提升 3 个数量级。
1. 计算参数导致的系统误差
泛函选择偏差:GGA 泛函(如 PBE)通常低估电荷转移量,而 HSE06 杂化泛函可更准确描述共价相互作用。在分析 CO₂吸附于金属有机框架(MOF)时,PBE 计算的 C-O 键电荷聚集量比实验值低 15%,而 HSE06 结果与 X 射线光电子能谱(XPS)偏差 。
基组不完全性误差:对含重元素(如 Au、U)的体系,需使用相对论赝势(如 Scalar-relativistic pseudopotential)并包含全电子芯态,否则会导致 5d 轨道电荷密度计算偏差 > 20%。
2. 可视化与量化分析的衔接问题
等值面阈值的主观选择可能导致误判:建议采用多个阈值(如±0.005、±0.01、±0.02 e/ų)对比分析,或结合电荷密度差分的空间积分(如某区域总电荷变化量)进行定量佐证。
三维数据降维带来的信息损失:对于复杂体系(如多组分催化剂),需结合三维等值面与二维切片的多角度分析,避免单一视角下的特征遗漏。
随着量子计算与高分辨率表征技术的发展,差分电荷密度分析正朝着“计算–实验” 协同验证方向演进。例如通过 4D-STEM(四维扫描透射电子显微镜)直接获取纳米尺度的电荷密度分布,与 DFT 计算的差分电荷密度进行空间匹配,可实现化学键形成过程的动态追踪。
同时,基于密度泛函紧束缚(DFTB)方法的实时差分电荷密度计算,将推动光催化、电催化等动态过程的原位分析,为揭示反应中间体的电荷演化规律提供关键工具。这种多尺度、多技术的融合,正使差分电荷密度分析从静态结构表征向动态机制解析跨越,成为连接量子理论与宏观材料性能的核心桥梁。