

看到新金属峰出现,就能直接写重构完成吗?
做电催化论文时,你一定见过这种表述:反应过程中原来的氧化物峰逐渐变弱,新的金属峰出现,所以催化剂发生了重构。
这个判断方向没错,但如果只靠一张原位XRD图就写“重构完成”,很容易过度解读。因为峰变弱可能来自晶粒变小、结晶度下降、无定形化、检测限变化;
金属峰出现也只能说明有金属相生成,不等于原来的母相完全消失,更不等于这个新相就是唯一活性相。


先说结论:
原位XRD峰变弱:说明晶态结构在反应中发生变化,但不能单独证明完全重构。
金属峰出现:说明有新金属相生成,但要区分“部分还原”和“完整工作态转化”。
判断重构要看证据链:原位相变 + 价态变化 + 配位切换 + 形貌重组 + 性能/机理关联。
最稳妥的写法是:在特定反应条件下,初始相逐步转化为某种工作态结构,而不是一句话写死“催化剂完全重构”。
催化剂重构要靠哪些证据链判断?
这一节先搭一张“相结构—价态—配位—形貌—性能”的重构判断地图。接下来的总览图重点看三个关键词:相变、价态、活性相。
原位XRD/Raman负责看相结构变化,XANES/EXAFS/XPS负责看价态和配位变化,TEM/SEM与性能数据负责确认重构后的结构是否真的参与反应。

什么是催化剂重构?
人话版:催化剂重构,就是材料在真正反应条件下“变样了”。
它可能从氧化物变成金属,也可能只是表面几层原子变了,还可能生成新的晶界、缺陷、应变、纳米颗粒或无定形层。
专业版:电催化重构指催化剂在外加电位、电解液、反应物、局部pH、电场和传质等作用下发生的相组成、价态、配位环境、形貌或表面化学状态演化。
重构可以发生在表面,也可以扩展到体相;可以是部分重构,也可以接近完整重构。

人话版:所以,重构不是一个简单标签。它更像一个过程:反应前的材料可能只是“前驱体”,真正干活的可能是反应中形成的新结构。
专业版:在很多电催化体系里,初始催化剂和工作态催化剂并不完全一致。
判断清单:判断活性相时,不能只看反应前结构,也不能只看反应后静态图,而要追踪反应过程中结构如何演化。
判断公式:催化剂重构 = 反应条件触发结构变化 + 初始结构信号衰减 + 新工作态信号出现 + 新结构与性能/机理相关。
适用边界:只看到表面变粗糙,不一定叫有效重构;只看到价态降低,也不能说明已经形成稳定工作态;只看到金属峰出现,也不能证明母相完全消失。
催化剂重构为什么重要?
人话版:因为很多论文里,反应前精心合成的材料,不一定就是反应时真正发挥作用的材料。真正决定选择性和稳定性的,可能是反应中长出来的金属颗粒、晶界、低配位位点或界面结构。
专业版:电催化反应通常发生在强电场、极端电位和复杂电解液界面中。
此时催化剂表面的热力学稳定性会改变,金属-氧键、金属-金属键、吸附中间体覆盖度和局部离子环境都会影响结构演化路径。
人话版:这也是为什么同样写“氧化物衍生金属”,有的容易产多碳产物,有的更容易产氢气,有的很快失活。差别可能不在反应前,而在反应中重构出了什么工作态。
专业版:重构后的结构可能改变关键中间体吸附,例如*H、*CO、*COH、*OCCOH等,也可能改变电荷转移阻力、局部pH和反应物供应。
因此,重构既可能带来活性提升,也可能导致结构坍塌、团聚和失活。
判断公式:重构是否重要 = 重构结构能被检测到 + 重构程度与性能趋势对应 + 重构位点能解释关键中间体和副反应变化。适用边界:不要默认“重构越彻底越好”。
有些体系需要部分保留氧化态界面,有些体系需要完全转成金属态,有些体系则依赖动态平衡态。好不好,要看证据链能不能闭环。
这些数据到底怎么看?
第一步看原位XRD或原位Raman。原来的峰是不是在反应中逐渐变弱?新峰是不是同步出现?如果只是反应后出现新峰,信息就少一半,因为你不知道它什么时候出现、怎么出现。
原位XRD主要追踪晶态体相或较大晶区的相变化,适合判断晶相衰减和新晶相生成;
原位Raman更敏感于表面或近表面振动模式,适合补充XRD看不到的表面结构演化。
第二步看价态。比如吸收边是不是向金属参比靠近?XPS里金属态比例是不是升高?但XPS偏表面,还容易受空气暴露影响,所以不能单独决定整体价态。
XANES可判断平均价态和局部电子结构;XPS可补充表面价态;但二者探测深度不同。若XANES接近金属参比,而XPS还有少量高价峰,需要考虑表面再氧化、空气暴露或外层氧化皮。
第三步看配位。原来的M-O配位是不是变弱或消失?新的M-M配位是不是增强?如果M-O还在,通常说明母相或氧化物环境并没有完全消失。
EXAFS可以看局部配位壳层。氧化物到金属态重构时,常见变化是M-O峰衰减、M-M峰增强,甚至M-O-M长程配位消失。若再结合WT-EXAFS,可以更好地区分不同散射路径。

第四步看形貌。反应后到底是生成颗粒、枝晶、薄层、晶界,还是团聚块?这一步决定你能不能把“重构”写成具体结构,而不是空泛一句话。
TEM、HR-TEM、SEM、EDS和FFT可以验证重构后的纳米结构、晶界、晶面、元素分布和相分离情况。
尤其当论文声称晶界、低配位或应变是活性位点时,显微结构证据非常关键。
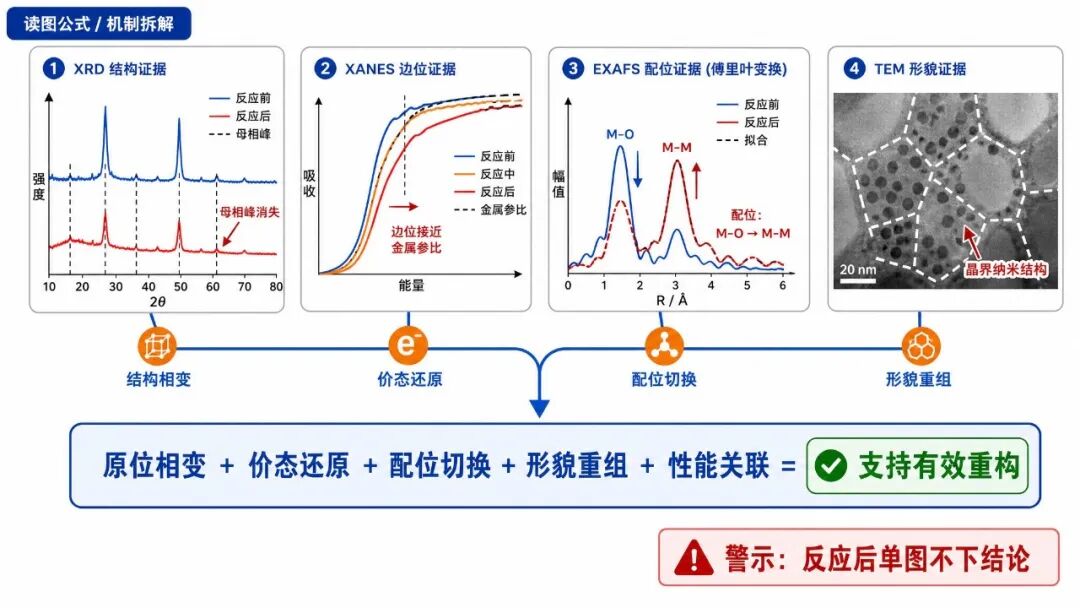
读图公式1:原位XRD母相峰变弱 + 新相峰出现 = 支持反应中发生晶相演化。
读图公式2:XANES边位接近金属参比 + XPS金属态比例升高 = 支持价态还原。
读图公式3:EXAFS中M-O配位减少、M-M配位增强 = 支持局部结构由氧化物环境转向金属环境。
读图公式4:TEM/SEM看到新纳米结构、晶界或颗粒 = 支持重构后形貌真实改变。
读图公式5:重构结构与FE、分电流密度、HER抑制、DFT能垒对应 = 支持该重构结构是功能相关活性相。适用边界:XRD看不到不等于绝对没有;
XPS看到高价峰不等于反应中没有金属态;TEM局部区域漂亮不代表全电极一致。最好让多种证据趋势互相对上。
什么时候不能这样判断?
人话版:如果只有反应后XRD,看到金属峰,就写“完全重构”,这很危险。因为反应后样品经历了取出、洗涤、干燥、空气暴露,结构可能已经不是工作时的真实状态。
专业版:ex situ表征只能提供反应后快照,无法说明重构发生的时间顺序、动力学快慢和反应中稳定性。判断工作态重构,原位或操作态证据更有说服力。
人话版:如果只看XPS,也容易误判。XPS看的是表面几纳米,表面还原不代表整体还原;表面氧化也不代表反应中没有金属态。
专业版:XPS受表面氧化、碳污染、峰拟合参数和探测深度影响。对于整体重构程度,XAS、XRD和显微结构证据通常更稳;对于表面价态,则XPS更敏感。
人话版:如果只看到性能变好,也不能倒推“一定是重构造成的”。性能提升可能来自面积增加、传质改善、局部pH变化、电导提升或测试构型差异。
专业版:性能数据需要与结构演化建立因果关联。仅有FE或电流密度提升,不足以证明重构结构是活性相;
必须结合中间体表征、动力学分析和理论计算,说明新结构如何影响反应路径。
判断公式:不能下强结论的情况 = 只有单张反应后图 + 没有原位过程 + 没有配位证据 + 没有形貌证据 + 没有性能/机理关联。
前面讲的是通用方法,下面用相关研究做一个垂直案例。
这个案例的价值,不是简单证明某个催化剂性能高,而是展示如何把“界面微环境改变—重构路径改变—工作态活性相形成—产物选择性提高”连成一条完整证据链。
回到相关研究:这个案例在验证什么?
这个案例要验证的问题是:电极结构能不能通过调控界面微环境,让初始氧化物在CO2还原过程中更快、更完全地转化成富晶界金属纳米结构,并进一步提高C2+产物选择性。
换句话说,它不是只问“有没有Cu0生成”,而是问四件事:重构为什么更快?重构是否更完整?重构后形成了什么结构?
这个结构为什么更有利于C-C偶联而不是HER或CH4路径?

结构/组成证据如何支撑主题?

通用判断规则:如果要证明重构差异来自界面微环境,而不是初始材料本身差异,第一步必须确认反应前样品是否可比,并证明结构设计确实会改变传质、局部pH和电场。
在这个案例中,研究者先用界面示意图提出三个控制重构的关键因素:表面电场、局部pH和传质效率。

FEM模拟显示,三维有序大孔结构能同时加快物质扩散、提高局部OH−浓度并增强表面电场。
孔连接处局部OH−浓度最高可到1.63 m,相比平面结构附近约1.11 m明显升高;表面电场也增强约88%。
这说明结构设计不是单纯增加表面积,而是在电极/电解液界面同时改变多个变量。
更快的CO2传输、更高的局部碱性和更强的电场,都可能促进氧化物还原、溶解-再沉积和金属Cu形成。
接着,反应前表征确认两个对照样品都属于相纯正交Fmmm结构。PXRD和Rietveld精修证明相结构一致;SEM/TEM显示一个样品具有三维连通大孔框架;

EDS证明La、Cu、O均匀分布;Cu K-edge XANES和EXAFS显示两者反应前Cu价态和配位环境接近。
能下的结论:反应前主要差异来自架构和界面微环境,而不是初始Cu电子结构或配位环境明显不同。
不能过度解读:FEM模拟提供的是趋势和空间分布预测,不等于直接测到真实反应中的每一个孔道pH和电场;反应前结构可比,也不代表反应中一定只有一个因素在起作用。
电荷/界面/动力学证据如何接上?

通用判断规则:如果界面微环境真的促进有效重构,性能上应该出现三个信号:目标产物FE提高、分电流密度增大、副反应HER受抑制。同时,动力学表征应显示界面反应阻力降低。
性能数据里,3DOM样品在−2.0 V vs. RHE时FEC2+达到70.4%,高于bulk样品和商业CuO。
在−2.6 V时,C2+分电流密度达到585 mA cm−2,也明显高于对照样品。更重要的是,高阴极电位下FEH2仍低于15%,说明HER被有效压制。
为了避免“只是面积更大”的误判,研究者还做了ECSA归一化。
归一化后,3DOM样品的C2+分电流密度仍然最高,说明性能提升不只是表面积贡献,也包含更优的本征/界面反应能力。
EIS和DRT进一步说明动力学差异。Nyquist图中3DOM样品半圆最小,说明电荷转移阻抗低;
DRT中频区对应CO2RR/HER耦合电荷转移过程,3DOM样品中频峰位更高频、阻力更低,说明界面反应动力学更快。
能下的结论:界面微环境调控带来了更高C2+选择性、更大C2+产率、更弱HER竞争和更快界面反应动力学。
不能过度解读:性能提升本身还不能证明重构路径。它只能说明结果更好,真正的重构证据必须回到原位XRD、Raman、XAS、EXAFS和TEM。
性能提升是否站得住?
通用判断规则:判断催化剂是否真的重构,要重点看“母相是否衰减、新相是否出现、价态是否还原、配位是否切换、形貌是否重组”。
如果这几类证据同时指向同一结论,可信度才高。
原位XRD显示,在CO2RR条件下,两个样品的初始氧化物特征峰都会逐渐衰减,并出现金属Cu0峰。但3DOM样品的相变明显更快,说明其重构动力学更快。
在高电流密度流动池中反应1 h后,ex situ XRD显示3DOM样品只剩金属Cu0衍射峰,而bulk样品仍保留初始相和金属Cu0共存。
这是区分完整重构和部分重构的核心证据:前者母相信号基本消失,后者仍有残余母相。
原位Raman也支持这一点。3DOM样品的初始相振动模式在3 h后消失,而bulk样品仍能检测到相关振动模式。
也就是说,3DOM样品中若还有残余初始相,其含量至少低于XRD/Raman检测限,不太可能作为主要活性相解释性能。

XAS进一步证明价态和配位切换。3DOM反应后样品的Cu K-edge XANES吸收边接近Cu箔参比,说明Cu基本还原为Cu0;
FT-EXAFS和WT-EXAFS显示其主要为Cu-Cu配位,初始氧化物框架相关的Cu-O-La配位消失。
bulk样品则仍同时存在Cu-Cu、Cu-O和Cu-O-La信号,说明重构不完全。
TEM给出最终形貌证据。bulk样品形成的是残余母相表面负载的Cu纳米颗粒;3DOM样品则变成自组装树枝状纳米Cu0结构,并含有丰富晶界。
HR-TEM还能直接看到两个Cu(111)晶畴之间的晶界区域。
EXAFS拟合显示3DOM重构后Cu配位数约7.27,Cu-Cu键长缩短到2.53 Å,对应约0.65%压缩应变。
能下的结论:3DOM架构驱动了更快、更完整的重构,使初始氧化物转化为富晶界、带中等欠配位和压缩应变的纳米Cu0工作态结构。
不能过度解读:XPS中仍有少量Cu2+峰,研究者将其归因于空气暴露后的表面氧化。
因此不能只凭XPS表面高价峰否定工作态Cu0,也不能只凭单张TEM图代表整个电极,需要和XRD、Raman、XAS、EXAFS共同判断。
稳定性和机理是否闭环?

通用判断规则:证明重构还不够,还要证明重构后的结构为什么有用。要问:它改变了哪个中间体?抑制了哪个副反应?降低了哪一步能垒?长时间工作后是否还能保持?
原位Raman先分析界面水结构。bulk样品在高阴极电位下自由水比例明显增加,而3DOM样品表面的自由水比例基本稳定。
结合产物分布,研究者认为自由水更容易促进HER,而较稳定的氢键水网络更有利于C2+生成相关的质子转移。
原位FTIR进一步看中间体。3DOM样品出现明显约1200 cm−1的*OCCOH峰,且没有明显*OCCO信号,支持*CO与*COH耦合形成C-C键的路径。
bulk样品则有更强的*COOH和*CH2O相关信号,说明其更容易走C1产物或CH4相关路径。
此外,3DOM样品约1390 cm−1的CO3²−峰更强,支持其界面局部pH更高。
DFT把结构和路径连起来。计算显示,Cu配位数降低会让d带中心上移,但低配位不是越低越好。
3DOM重构后接近7.27的中等欠配位Cu更倾向稳定*CO2而不是*H,有助于抑制HER;bulk中更低配位的Cu位点更容易吸附*H,因此H2更多。
对于C2+路径,晶界和0.65%压缩应变可以稳定关键*OCCOH中间体,并降低*CO质子化为*COH的能垒。
也就是说,富晶界Cu原子不是简单的形貌描述,而是被中间体证据和自由能计算共同支持的潜在活性位点。
稳定性方面,MEA中样品在600 mA cm−2下稳定运行约200 h,并保持约70%的FEC2H4,FEH2低于15%。
长时间测试后,XRD和SEM仍显示其保持富晶界纳米Cu0结构,没有明显颗粒团聚。
能下的结论:重构后的富晶界纳米Cu0与界面水结构、中间体路径、DFT能垒和长时间稳定性形成闭环,因此可以被认为是与C2+生成密切相关的工作态活性结构。
不能过度解读:DFT模型是简化晶面和晶界模型,不能代表真实电极中所有位点;FTIR峰也可能有重叠和环境依赖。
更准确的说法是“多种证据共同支持晶界Cu位点促进C-C偶联”,而不是“唯一活性位点被绝对证明”。
最容易踩的三个坑
看到金属峰出现,就写“完全重构”。金属峰出现只能说明新金属相生成。
是否完全重构,要看母相XRD/Raman是否消失、EXAFS中M-O配位是否消失、XANES是否接近金属参比。
只看反应后表征,不看原位过程。反应后样品可能受到空气暴露、洗涤、干燥影响。判断工作态重构,最好有原位XRD、原位Raman或操作态XAS支撑。
把性能提升直接归因于重构。性能提升可能来自面积、传质、局部pH、电场或测试构型。要证明重构结构是活性相,必须连接中间体、能垒和副反应抑制。
认为低配位越低越好。这类体系里,中等欠配位可能更有利于目标路径;过低配位可能更容易吸附*H或走其他副反应路径。
下次读论文怎么判断?
先看原位相变:母相XRD/Raman峰是否随反应时间衰减?新相峰是否同步出现?
再看重构程度:反应后是否还保留母相峰、振动峰或M-O-M配位?是部分重构还是完整重构?
再看价态:XANES边位是否接近金属/低价参比?XPS是否一致?是否考虑空气氧化?
再看配位:EXAFS中M-O、M-M、M-O-M配位如何变化?有没有拟合配位数和键长?
再看形貌:TEM/SEM是否显示新的颗粒、晶界、枝晶、团聚或相分离?EDS能否说明元素分布?
再看性能关联:重构更快或更完整的样品,目标产物FE、分电流密度和ECSA归一化活性是否同步更好?
再看机制闭环:原位FTIR/Raman和DFT是否说明新结构改变了中间体、限速步或副反应?
最后看稳定性:长时间运行后工作态结构是否保持?有没有团聚、相变或活性位点流失?
记住一句话:判断催化剂重构,不能只问“有没有新峰”,而要问“母相是否消失、价态配位是否切换、形貌是否重组、性能和机理是否闭环”。