本文华算科技基于近年理论计算与实验研究进展,系统阐述自旋态对催化活性位点电子结构、反应路径及能垒的调控机制,重点解析典型计算模型的应用案例与构效关系研究方法,并通过关键图表揭示自旋极化对反应动力学的深层影响。
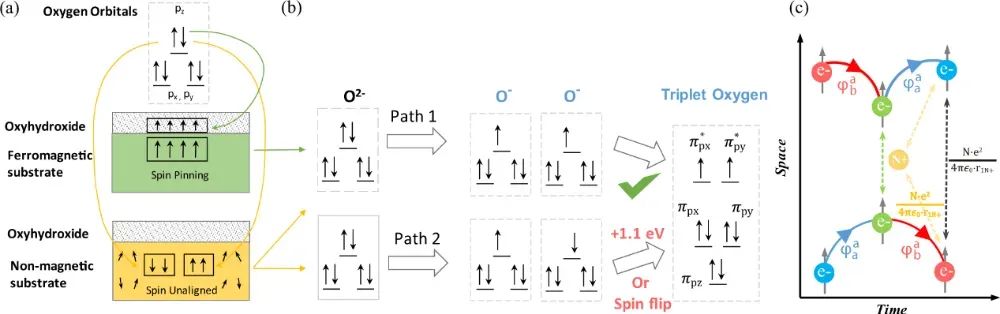
自旋状态通过改变催化剂活性中心的电子占据和轨道耦合,直接影响中间体的吸附行为与电荷转移路径。在OER四电子反应路径中(2H₂O→O₂+4H⁺+4e⁻),自旋极化通过三重态氧(³O₂)的生成/转化步骤显著影响能垒。
理论计算表明,基态氧分子为三重态(自旋多重度S=1),而反应物H₂O为单重态(S=0),自旋守恒要求催化过程需解决自旋翻转问题。
以LiCoVO₄为例,其反铁磁有序结构中存在沿Co-O键的磁极化通道,在施加电位时该通道可选择性提取特定自旋方向的电子,使界面累积磁矩,促进³O₂生成。
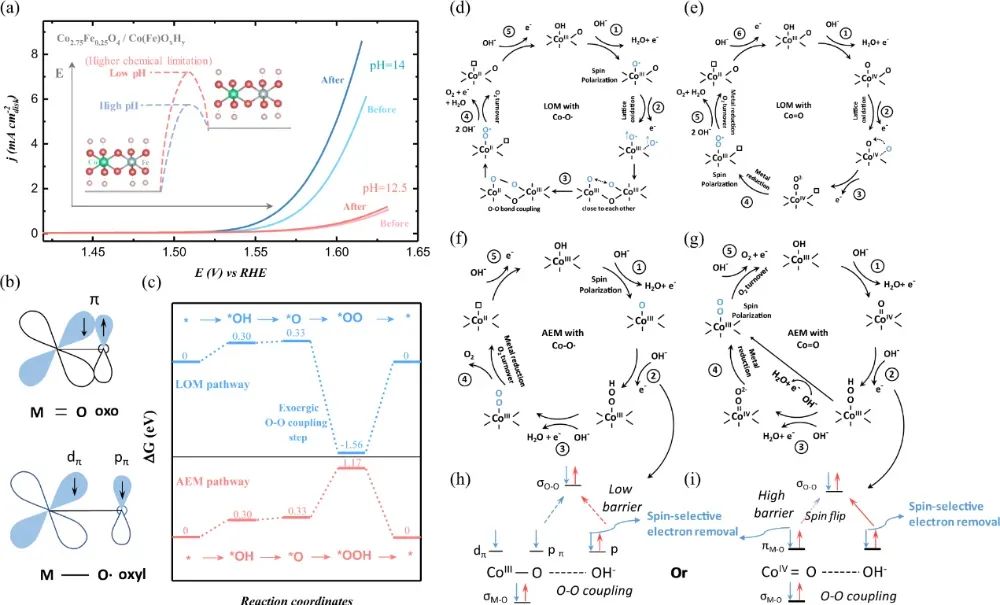
密度泛函理论(DFT)吉布斯自由能计算进一步揭示:自旋极化优化可降低速率决定步骤(O-O偶联)的能垒,如高自旋态CoOOH的OER过电位(226 mV)比低自旋态低148 mV。
DOI:
10.1038/s41467-021-23896-1
对于ORR过程(O₂+4H⁺+4e⁻→2H₂O),磁性催化剂中的交换相互作用可构建自旋过滤通道。
Pt₂Gd合金的DFT计算表明,Gd的4f轨道形成”反向自旋对”,削弱Pt-5d轨道自旋极化,使Pt位点d带中心上移,优化*OH吸附能。这种自旋屏蔽效应将ORR质量活性提升至1.5 A·mg⁻¹,半波电位达0.95 V。自旋调控还通过手性诱导自旋选择性(CISS)效应影响反应路径,手性金属氧化物可通过不对称电子传递实现自旋极化电流,降低OER/ORR过电位。
自适应自旋约束DFT方法突破传统计算限制,可处理非共线磁性构型与自旋–轨道耦合效应。以单层CrI₃为例,该方法通过约束磁矩幅度和旋转角度,捕捉自旋波动导致的带隙变化:当自旋构型从铁磁态(↑↑↑)转变为120°非共线态时,费米面附近出现新的自旋极化能带,dₓy轨道分裂能增加0.8 eV。
该模型结合机器学习可高效生成自旋-晶格相图,为催化剂设计提供高精度数据库。
DOI:
10.1038/s41467-021-23896-1
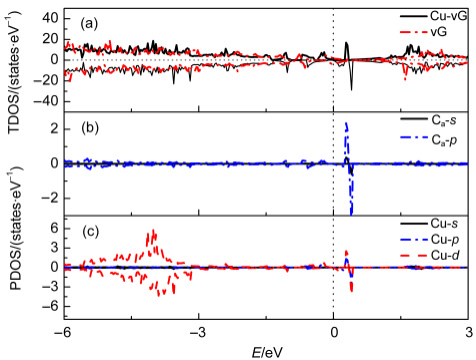
自旋分辨态密度(PDOS)分析是揭示电子结构调控的核心工具。Pt₂Gd合金的PDOS显示:Gd-5d轨道在-2.5 eV处形成自旋不对称峰(↑↓电子数比1.7:1),而Pt-5d轨道在费米能级处的自旋极化率从纯Pt的32%降至18%,证实自旋屏蔽效应。类似地,缺陷石墨烯负载Cu单原子的PDOS中,Cu-3d轨道在-1.8 eV处出现明显自旋分裂峰(↑态密度峰值比↓高40%),增强*OOH中间体的吸附强度。吉布斯自由能图直观展示自旋态对反应路径的影响。OER四步反应中(OH→O→OOH→O₂),高自旋Co³⁺的O→OOH步骤ΔG为1.52 eV,显著低于低自旋态的1.89 eV。关键源于e_g轨道电子占据:高自旋态(t₂g⁴e_g²)的e_g电子通过σ键与氧中间体强耦合,而低自旋态(t₂g⁶e_g⁰)缺乏此作用。双原子催化剂(DACs)的DFT计算进一步揭示,Fe-Co双位点的反铁磁耦合使自旋通道导通,将O质子化能垒从0.75 eV降至0.48 eV。DOI: 10.1038/s41467-021-22865-y
自旋交叉能垒计算采用多参考态方法(CASSCF)处理Fe-S簇。在[Fe₃S₄]立方烷中,活性位点自旋态(S=2/5/2)间能差仅1 mHa,几何畸变可使高自旋态稳定性提升3.2 kcal/mol。这种亚稳态特性使自旋态易受配体场调控,为动态反应提供基础。
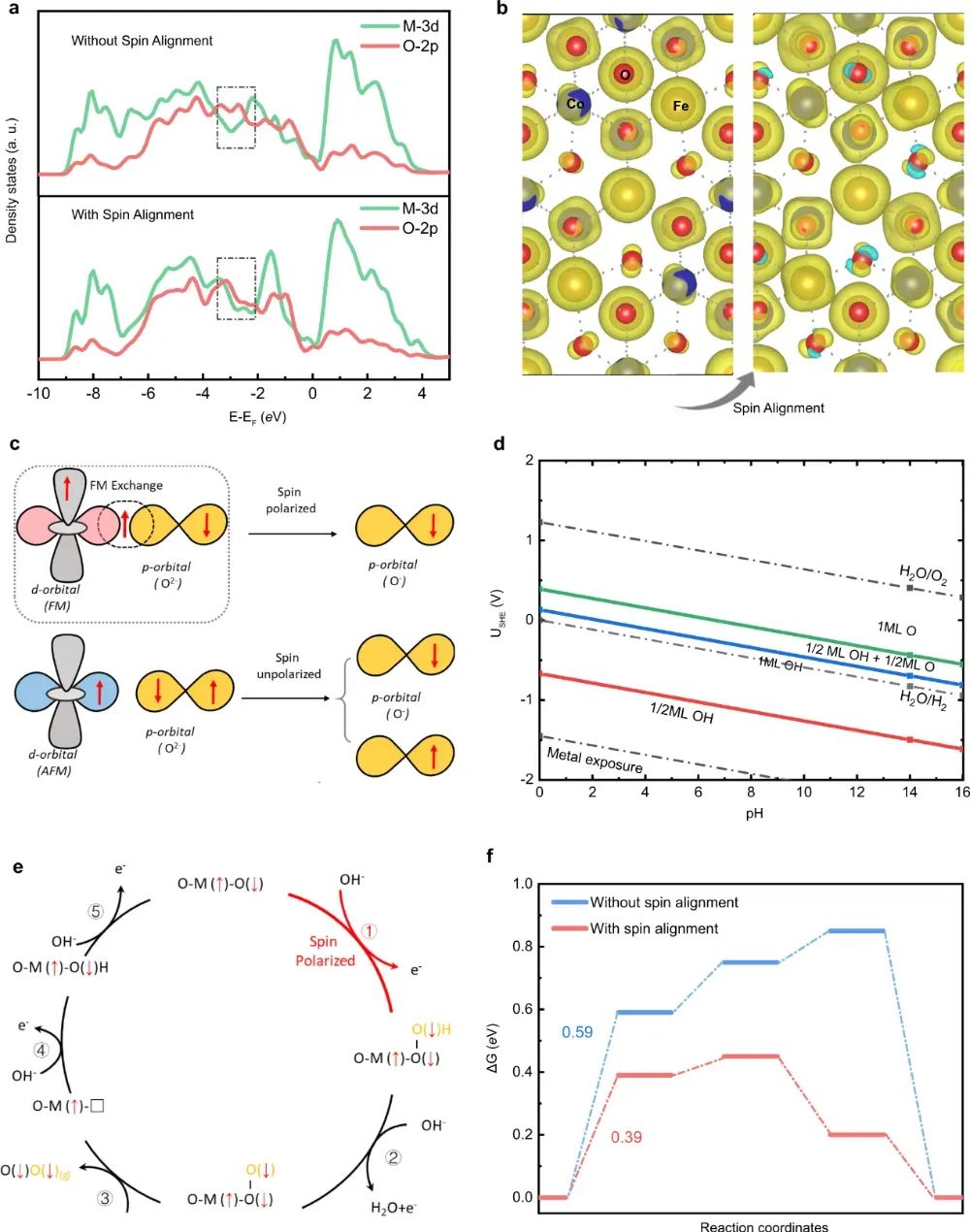
结构重建策略:Co₀.₈Mn₀.₂ MOF经磁刺激重构后,Mn³⁺诱导Co²⁺从低自旋(LS)向高自旋(HS)转变。XAS证实Co-L₃边白线强度增加2.3倍,表明未配对e_g电子增多;OER质量活性达3514.7 A·g⁻¹,DFT计算显示HS态使*OOH解吸能降低0.27 eV。
类似地,五水铁矿经十二烷基硫酸钠修饰后,Fe³⁺自旋态从S=1/2(LS)转变为S=5/2(HS),Mössbauer谱显示四极分裂值ΔE_Q从0.38增至0.92 mm/s,对应八面体对称性破缺。
DOI:10.1038/s41467-021-22865-y
外部磁场效应:LaCoO₃在0.5 T磁场下OER活性提升400%,原位磁圆二色谱显示Co³⁺的HS组分从18%增至65%。微动力学模型结合DFT证明:磁场促进自旋极化电子转移,使O-O偶联活化熵减少12.7 J·mol⁻¹·K⁻¹。
同步辐射技术:IrSA-Pt NPs催化剂的XANES谱显示,Ir-L₃边能量比金属Ir低1.3 eV,表明电子从Pt转移至Ir;EXAFS拟合证实Ir-O配位数从6.0降至4.2,形成配位不饱和位点。DFT计算表明,这种重构使Ir-5d轨道自旋极化率提高至42%,优化*O吸附强度。
自旋极化STM:NiFeOx催化剂表面观测到自旋相关电子隧穿,在+1.2 V偏压下↑自旋电子隧穿概率比↓高3倍,直接验证磁性通道存在。结合ΔΨ理论模型(自旋分辨功函数差),证实自旋极化率与OER过电位呈负相关(R²=0.91)。
动态自旋追踪:AIMD模拟显示,800 K时Al₂O₃表面氧空位迁移率增加,导致Fe活性中心自旋态在LS/HS间震荡(震荡周期)。需开发含时自旋DFT方法捕捉瞬态过程。
多自旋态耦合机制:Fe₃O₄的QMC计算表明,界面存在S=1/2(Fe²⁺)-S=5/2(Fe³⁺)自旋对,交换积分J=-15 meV驱动自旋翻转。需建立多体波函数描述符关联自旋序与反应能垒。
异质界面自旋输运:Pt/Gr/MoS₂异质结的NEGF计算揭示,石墨烯层产生自旋过滤效应(↑透射率92% vs ↓透射率37%),但界面散射导致自旋弛豫时间。需设计自旋保护层维持长程相干性。
未来方向包括:开发机器学习自旋势场(如DeepH方法预测磁矩分布);利用X射线磁圆二色(XMCD)成像技术实现单原子自旋态原位测绘;设计双原子自旋催化位点(如Fe₂-N₆)实现三重态氧定向转化。
自旋调控通过改变活性中心电子占据、轨道耦合及磁交换作用,为突破OER/ORR能垒极限提供新路径。
理论计算不仅揭示自旋极化态密度、自旋通道导通、自旋交叉等微观机制,更与实验表征形成闭环验证。随着自旋分辨表征技术和多尺度模拟方法的融合,自旋工程将推动下一代高效氧电催化剂的设计范式革新。
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!