

Q1:朱老师,可以取周期性结构的重复单元放在晶胞里做吸附计算吗?
A:这样不再是周期性结构,只能用0维来处理,最好用高斯计算
Q2:朱老师,开始算了一点点然后计算就停了,出现这个报错,怎么解决呀,才算了6步
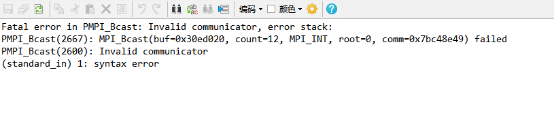

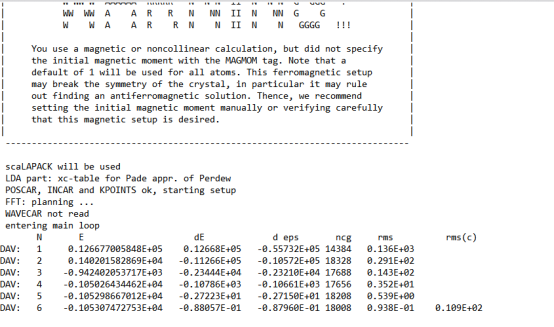
A:换个algo试试
Q3:朱老师,我想问下自旋向上和自旋向下的dos图不对称时,那个d带中心是怎么算的?
A:分开算,也有算完用平均值的
Q4:朱老师,我将晶体中的Fe-N键换成了Ni-N键,发现Ni-N键使得体系的形变减小,该用什么分析描述这个结果呢?
A:用bader电荷或者态密度
Q5:朱老师,bader电荷计算结果显示表面原子得到电子变多,吸附增强,那么d带中心应该是上移吗?或者说是原子得电子d带中心应该怎么变化呢?
A:这个不一定,但是一般来说价态越低吸附越强
Q6:朱老师,VASP计算的mag=11.6370就是计算出来的磁性吧,那找个单位是μB吗?
A:是的
Q7:朱老师,过渡金属的初始磁矩设置多少呢?
A:5
Q8:朱老师,计算ci-neb过渡态的时候,一般对于过渡态,EDIFFG的设置要到多少啊?
A:-0.03左右
Q9:朱老师,1t’相结构的二维材料,超胞需要切面找001面吗,beta角93.91,不切面的话会影响后续HER计算吗
A:不会,结果一样
Q10:朱老师,审稿人对我的cohp图提出了异议,让我重新算算确认其准确性,上图右边是态密度,左边是其cohp图,有什么问题呢?It is suggested that the author recalculate the ICOHP value in Fig. 8 to confirm the accuracy
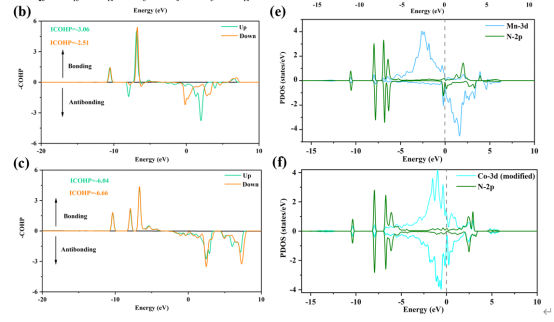
A:提高vasp的k点试试
本次答疑由拥有15年VASP实战经验的华算科技朱老师(同济大学本博、深圳海外高层次人才)提供。?点击进入《VASP | 理论计算常见问题解答专题》,快速查找更多计算解决方案。