
VASP中初始结构不做频率计算,Gaussian中每一步都做频率计算,所以Gaussian计算的初始结构比VASP多一个自由能校正值,这也导致了VASP计算出来的吸附过程是吸热的,而Gaussian算出来是放热的
原因1:对于周期性体系固体表面吸附的自由能计算来讲,近似认为固体表面能量受温度影响较小,不做频率矫正。
原因2:孤立分子体系的自由能用高斯计算更为准确,需要每一个结构都计算频率,因此用VASP和Gaussian进行对比并不合适。
Q3:朱老师,差分电荷密度盒子角上出现这种应该是通过哪个命令调高计算精度?
检查角上有没有原子,如果没有的话可以进行如下调整;
首先检查每个计算是否收敛,没有收敛的话重新计算至收敛;
其次检查电荷密度差的减法,需要为AB-A-B。
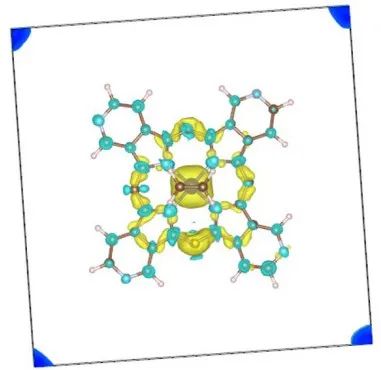
Q4:朱老师,得消除层间的相互作用才能导出正确的电子密度吗?
Q:老师,因为我的体系中含有Br,初始结构优化的时候层间的Br像是成键了,直接导出的电子密度是错误的,所以得消除层间的相互作用才能导出正确的电子密度吧
结构优化会将对应原子弛豫到能量的稳定状态,不论结构优化之后原子之间是否靠近成键,需要通过结构是否散架来判断收敛的合理性而非通过想象的,所以,以优化后的结果为准。
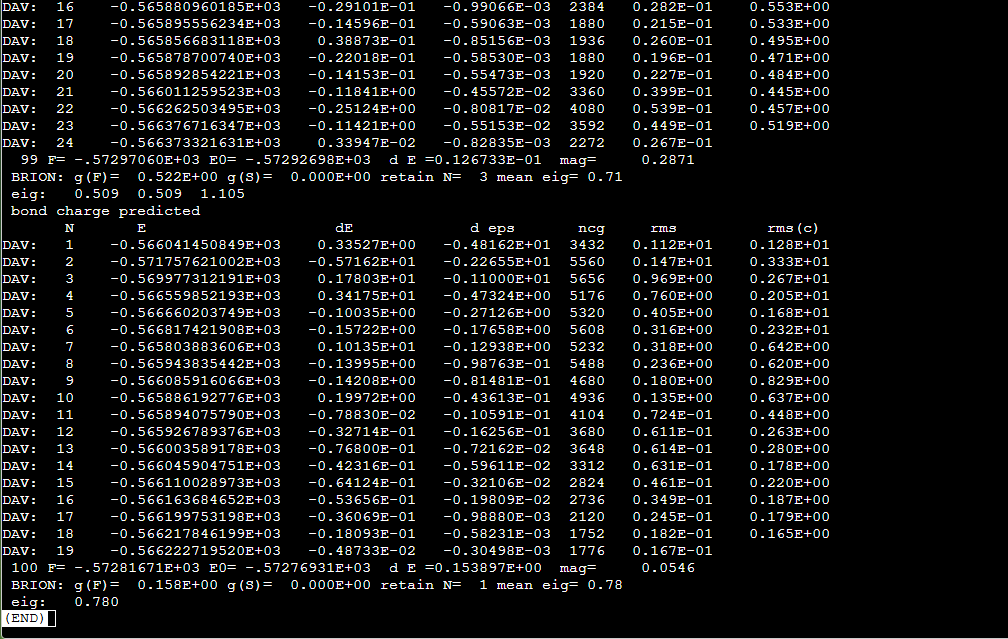
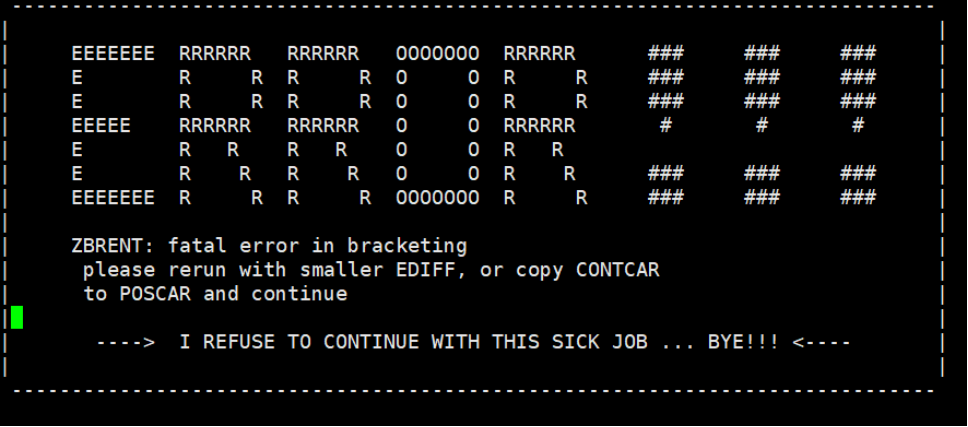
Q5:朱老师麻烦您帮我看看,这个有问题吗,不知道什么时候收敛?
如果需要使用nebef.pl脚本并且读取结构受力,必须先编译VTST到对应VASP程序里面:
搜索VTST进入官网,下载对应脚本文件上传服务器并解压使用;
将VTST算法在安装VASP的时候编译进去,可让超算工作人员处理;
过渡态计算本身时间较久难以收敛,可通过多次手动调整结构加快收敛速度。
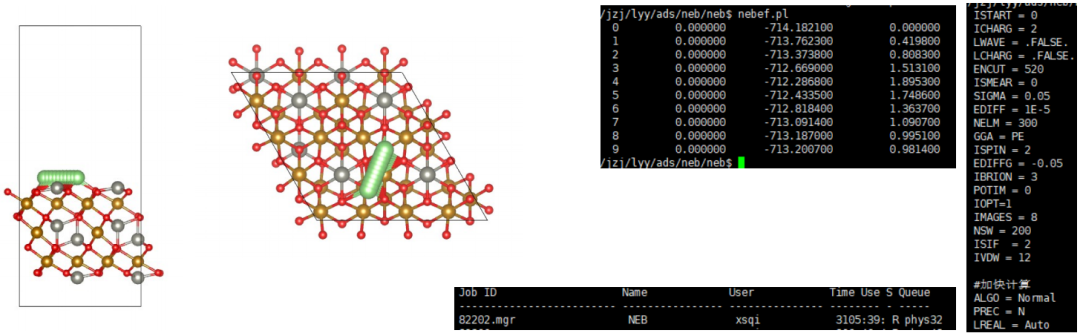
Q6:朱老师,衡量OOH与基底的成键强度的时候,可不可以用这三个金属-氧键的icohp的加和呢?
Q:老师您好,请问如果我的OOH*与催化剂基底上的两个Pt,Ni都形成了金属-氧键,那么我在衡量这个OOH与基底的成键强度的时候,可不可以用这三个金属-氧键的icohp的加和呢
COHP计算只能衡量单个化学键的键强,如果小分子共同吸附在3个原子上面,需要一个一个化学键进行计算,然而如果需要衡量这个OOH与基底的成键强度的时候…
对,所有的ICOHP数值加起来。
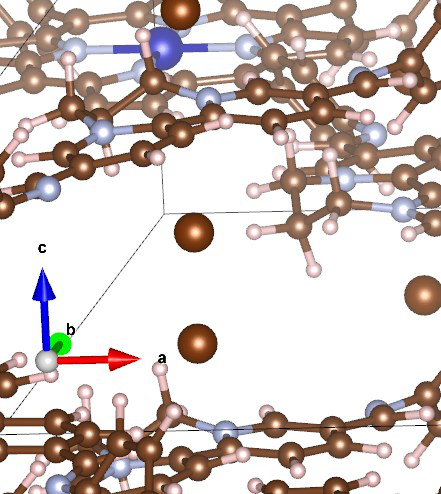
Q7:朱老师,对于晶胞边界有金属的情况,是不是会对磁矩的计算有影响呢?
在这里,需要先强调一点,在VASP计算中,结构都是周期性的,所以计算只取一个单胞内的原子进行计算,其他的做边界近似处理,因此:
关注单胞内原子的位置是否位于单胞中心其实没有必要;
在VASP计算中,所有的原子都是包含在内的,在保持晶胞参数不变的情况下,完全可以拖动整体结构将边角的金属原子放在中心;
所以,它位于边角并不会对磁矩计算有任何影响。
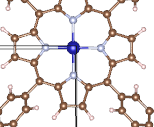
Q8:朱老师您好,想问下这个错误怎么解决,电子步运行了几步停了?
调节AMIX参数,增大和减小;
更换ALGO算法,建议用Fast;
关闭磁性,ISPIN=1。
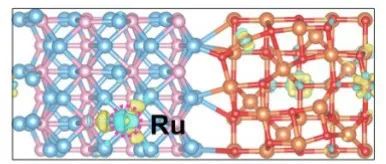
Q9:左边的Fe3O4上有很多的原子上有电荷密度,而右边的Ni2P上的原子却没有电荷?
Q:老师,我在求Ru单原子的差分电荷密度时,发现左边的Fe3O4上有很多的原子上有电荷密度,而右边的Ni2P上的原子却没有电荷,这样的结果是不是有问题,为什么会导致这个结果呢,我要如何调整呢。计算方法也是Ru=Ru/Ni2P/Fe3O4减去Ru再减去Ni2P/Fe3O4)
出现这个问题是因为磁矩在去掉Ru后发生变化,就会有电荷云;
计算时可以复制结构优化出来fe的磁矩,写到magmom里面;
INCAR加lorbit,outcar最后有每个原子磁矩。
Q10:没有出现reached required accuracy,是不是表示跑完了100步还是没有收敛?
在这里,需要先强调一点,在VASP计算中,所有结构优化的计算,正常收敛之后都会出现“reached required accuracy”:
INCAR参数里面NSW设置了每个结构优化计算的最大步长;
在VASP计算中,收敛完成计算即会结束,并出现“reached required accuracy”,通常不会达到最大步长;
所以,这个是没有收敛的。