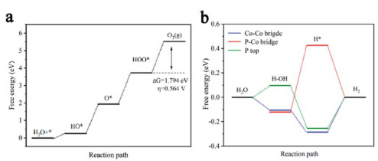
在氢气析出反应(HER)中,密度泛函理论(DFT)计算是研究催化剂性能和反应机理的重要工具。通过DFT计算,可以揭示催化剂表面的电子结构、吸附能、反应路径和能量变化,从而评估其催化活性和稳定性。

通过DFT计算比较CoC2O4、CoC2O4@MXene和R-CoC2O4@MXene三种材料在HER中的性能。图a和b展示了不同材料在不同能量状态下的电子分布,表明R-CoC2O4@MXene具有更优的电子结构。图d详细比较了这三种材料的吸附能,R-CoC2O4@MXene的吸附能最低,表明其具有更高的HER活性。
图e和f进一步展示了水分子在不同材料上的吸附自由能和氢气还原反应的自由能变化,R-CoC2O4@MXene在反应过程中表现出更低的能垒,说明其在HER中的稳定性更高。图g则通过示意图展示了水分子在R-CoC2O4@MXene表面的吸附、解离和氢吸附/解离的反应机制,进一步解释了其优异的HER性能。
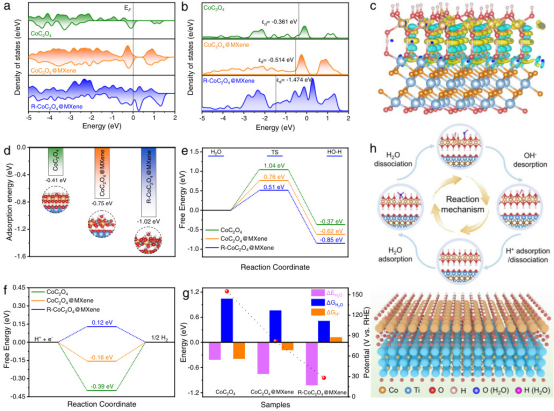

通过DFT计算比较了Sn3O6H6/MXene和MXene在HER中的性能。图A展示了吸附氢(Had)吸附、水吸附和水解离的能量计算结果,表明Sn3O6H6/MXene在水解离过程中具有更低的能垒。
图B和图C分别展示了水分子(初始状态,IS)和吸附氢(最终状态,FS)的优化配置,Sn3O6H6/MXene在反应路径中表现出更优的吸附和解离行为。图D和图E展示了CO₂-RR到HCOOH反应过程中的关键中间体Had+CO₂和OCHO,表明Sn3O6H6/MXene在HER中表现出更高的选择性。
图F和图G则展示了HER反应过程中的关键中间体H_ad和H₂,Sn3O6H6/MXene在反应过程中表现出更低的能垒,说明其在HER中的稳定性更高。
通过DFT计算研究了NiSe₂到NiSe的相变过程对HER性能的影响。NiSe₂在反应过程中逐渐转变为NiSe,Se原子向Ni原子的电荷转移导致电导率提高,d带中心向上移动,从而提高了HER性能。
通过DFT计算研究了Ni/TiO2-VO NFFs在HER中的性能显示了Ni/TiO2-VO NFFs具有更优的氢吸附吉布斯自由能(ΔGH*),在1 mol/L KOH溶液中,Ni/TiO2-VO NFFs在10 mA/cm²电流密度下的过电位为67 mV,且具有优异的稳定性,还展示了Ni/TiO2-VO NFFs在不同电势下的电流密度曲线,表明其在HER中表现出更高的催化活性。
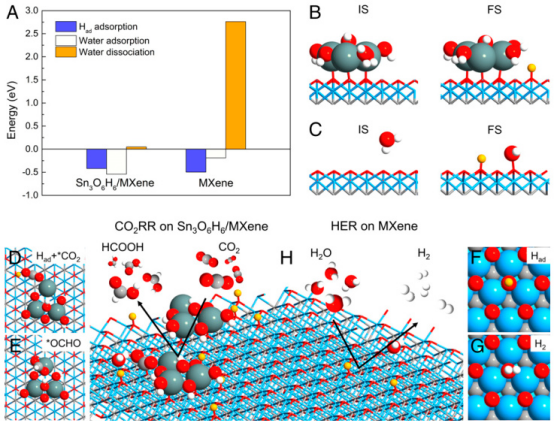
通过DFT计算研究了V0.2Mo0.8N1.2在HER中的性能。图显示了V0.2Mo0.8N1.2在不同电势下的电流密度曲线,Pt/C作为对比基准,V0.2Mo0.8N1.2表现出优异的催化活性。图还展示了这些催化剂的塔菲尔斜率与对数电流密度的关系,表明V0.2Mo0.8N1.2具有最低的塔菲尔斜率(39 mV dec⁻¹),显示出其优异的HER性能。图进一步展示了V0.2Mo0.8N1.2在0.5 M H₂SO₄溶液中长时间运行的稳定性测试,表明其在实际应用中的耐久性和可靠性。
通过DFT计算研究了CoP纳米粒子/碳纳米片在HER中的性能。图显示了CoP在不同晶面上的HER性能,表明CoP(100)和Co2P(121)表面具有较大的放热自由能值,而CoP(112)@C和CoP(211)@C的自由能值则较大且吸热。图还展示了引入N掺杂剂可以降低CoP(112)@NC和CoP(211)@NC的自由能值,提高HER性能。图进一步展示了CoP(100)@NC的自由能值最低,表明其具有最佳的HER活性。