能带结构是固体物理和材料科学中的一个核心概念,它描述了固体中电子的能量分布情况。能带结构的计算对于理解材料的电子性质(如导电性、光学性质等)至关重要。
能带结构是指在周期性晶体中,电子的总能量随波矢 k 的变化而形成的能量分布。在固体中,由于电子之间的相互作用,单个原子的能级会分裂成多个能带,这些能带之间可能存在能量间隙(即带隙)。能带结构的计算通常基于密度泛函理论(DFT)。
首先需要对材料的晶体结构进行优化,以获得稳定的晶格参数。这一步骤可以通过自洽场(SCF)计算来完成。
在结构优化完成后,进行静态自洽计算,以获得体系的波函数和电荷密度。这一步骤是计算能带结构的基础。
在静态自洽计算的基础上,进行非自洽计算,以计算能带结构。这一步骤通常使用bands命令进行。
对计算结果进行后处理,包括重新排序能带、计算能带相关属性(如带隙、费米能级等)。
使用plotband命令绘制能带图,展示能带结构。
VASP:通过vaspkit生成KPOINTS文件,进行能带计算。
PWSCF:通过relax、scf、nscf、bands等命令进行能带计算。
GPAW:通过fixed_density设置kpts路径,进行能带计算。
半导体材料的能带结构图中横轴表示沿着晶体结构中的特定方向(Γ-X-W-L-Γ-K-X),纵轴表示能量(以Rydberg为单位)。图中有多条曲线,代表不同能带的能量分布。在图的中间部分,有一个标记为“Fermi level”的区域,表示费米能级的位置。费米能级是电子在绝对零度下占据的最高能级,对于半导体材料来说,它决定了材料的导电性质。
声子晶体的能带结构图中分为两个区域:左侧为原子状态,右侧为固体状态。在原子状态中,能带是分离的,每个能级对应一个能带,能量水平相对较高。而在固体状态中,能带发生了重叠,形成能带结构,能量水平较低。图中阴影部分表示电子在不同能级上的分布情况,显示了电子在固体中的填充情况。
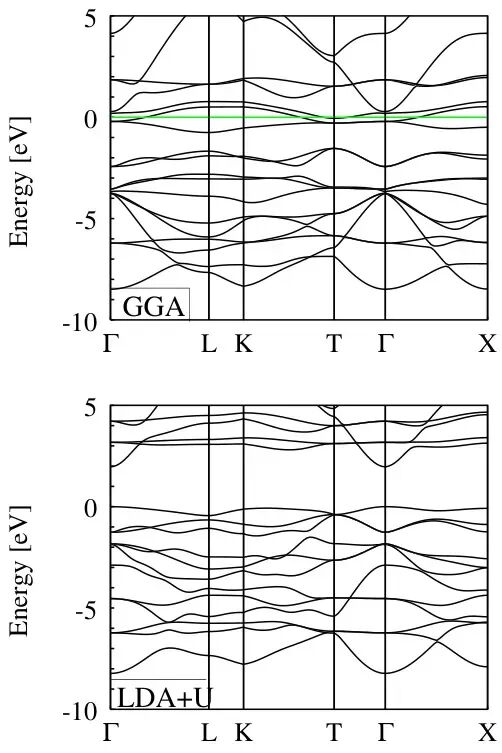
FeO材料在不同计算方法下的能带结构中上图使用GGA(广义梯度近似)方法计算得到的能带结构,而下图则使用了LDA+U(局部密度近似加上Hubbard U参数)方法。横轴表示k点路径,从Γ点到L点、K点、T点再到Γ点,最后到X点,纵轴表示能量(单位为电子伏特)。两条曲线分别代表了不同计算方法下FeO材料的能带结构,展示了不同方法对材料能带的影响。
在计算能带结构时,需要选择合适的高对称点路径。常见的高对称点包括Γ、X、W、L、K等。可以通过不同的软件和方法生成高对称点信息。
k点的采样数量和分布对能带结构的精度有重要影响。通常,k点的采样数量越多,能带结构的精度越高。
在计算能带结构时,选择合适的泛函(如PBE、GGA等)对结果有重要影响。不同的泛函可能会导致不同的能带结构结果。
在计算能带结构时,考虑自旋轨道耦合(SOC)可以更准确地描述材料的电子结构。特别是在过渡金属化合物中,自旋轨道耦合对能带结构有显著影响。
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!