

在能源转换与存储领域,氢气的析出反应(HER)和氧气的析出反应(OER)是电解水制氢的关键步骤,而MoS₂作为一种重要的二维过渡金属硫化物,因其独特的物理化学性质而备受关注。理论计算在研究MoS₂的HER和OER性能中发挥着至关重要的作用。通过计算催化剂的形成能,可以判断其稳定性和反应活性;计算反应自由能则能明确反应路径和能量变化;此外,还能通过模拟不同掺杂、应变等条件下的电子结构,揭示其催化机理。这些理论计算不仅为实验研究提供了理论指导,还为设计高效催化剂提供了新的思路和方向。接下来为大家提供几个具体的计算案例。
通过计算二维结构中吸附位点的电子轨道态密度,分析d带中心可以探究位点对OER/HER中间体的吸附强弱。
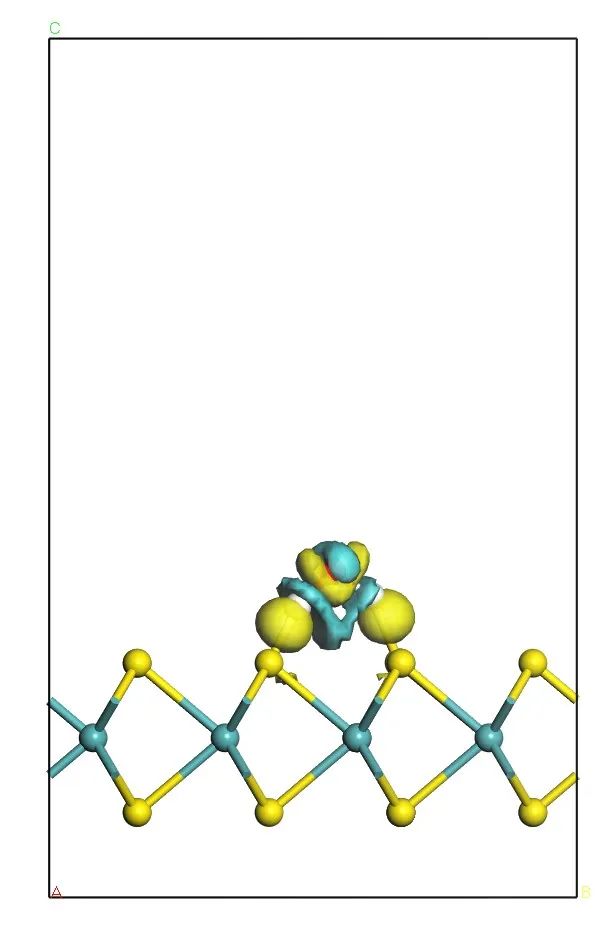
二、差分电荷密度图
对位点与吸附物质做差分电荷密度图,可以定性分析吸附中间体的电荷转移趋势,有利于分析吸附质与活性位点之间的相互作用。

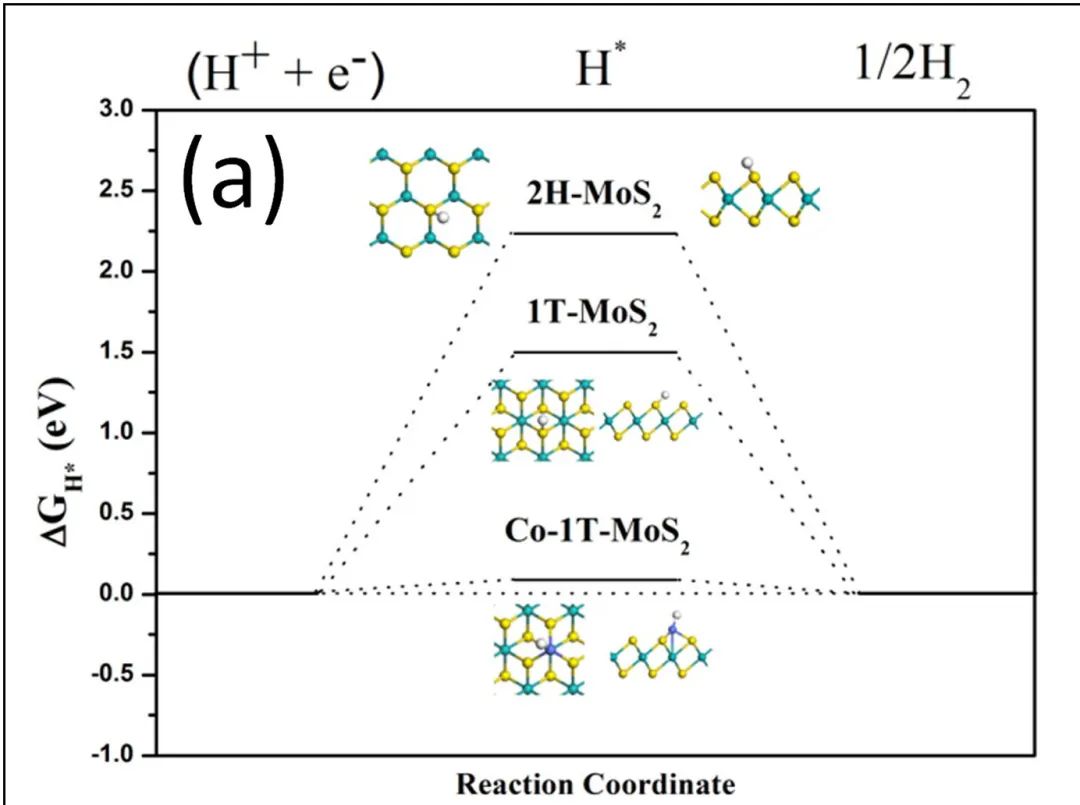
三、自由能台阶图
自由能台阶图可以得到催化反应过程中中间步骤的自由能变化以及对比不同催化剂的催化活性等。
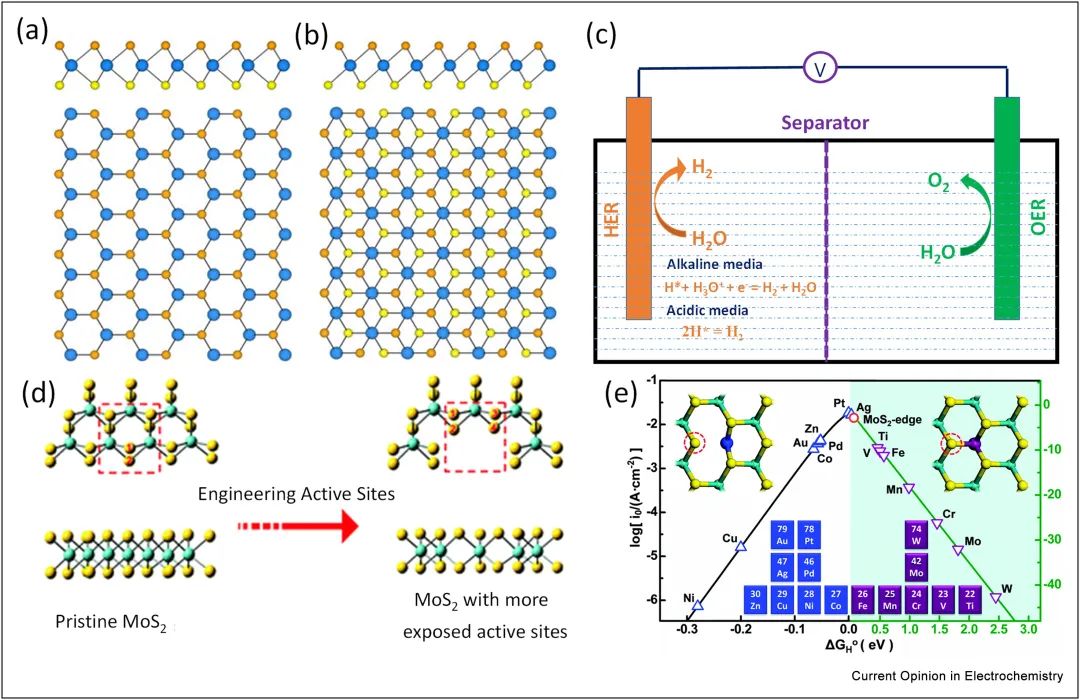
四、火山图
将*H中间体吸附自由能变差值与催化活性相关联可以得到火山图,直观看出大量不同材料的催化性能优劣并揭示中间体吸附强度与催化活性之间的关联。
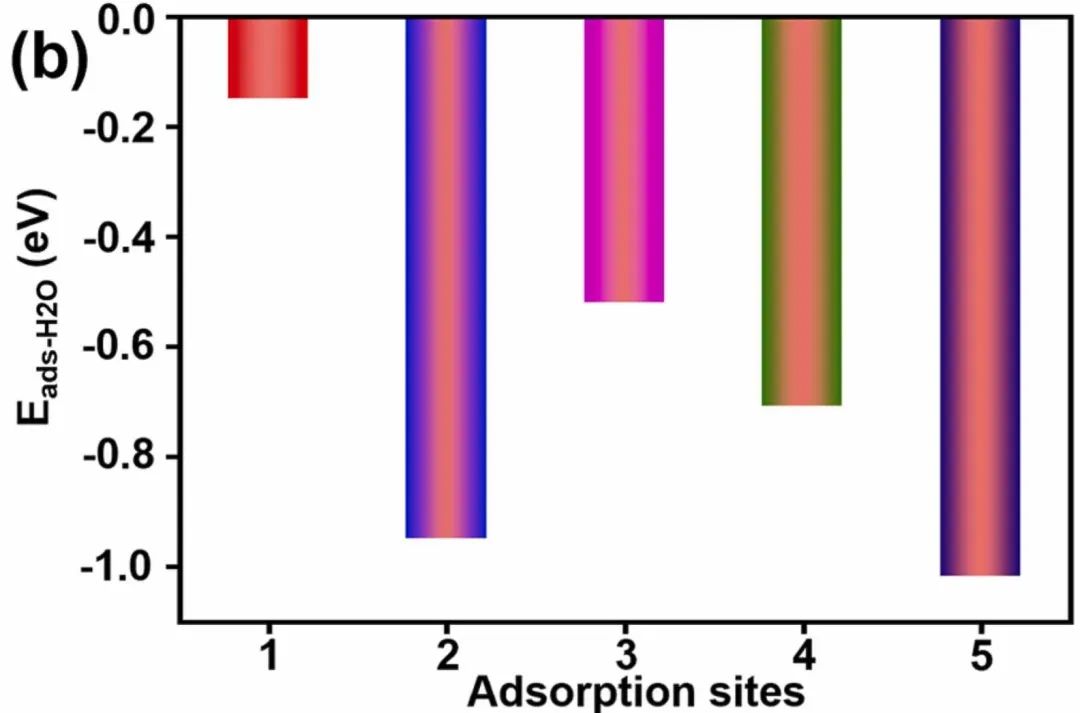
五、吸附能
吸附能可用于研究中间体在催化剂表面的吸附行为,有助于理解化学反应的催化机制。
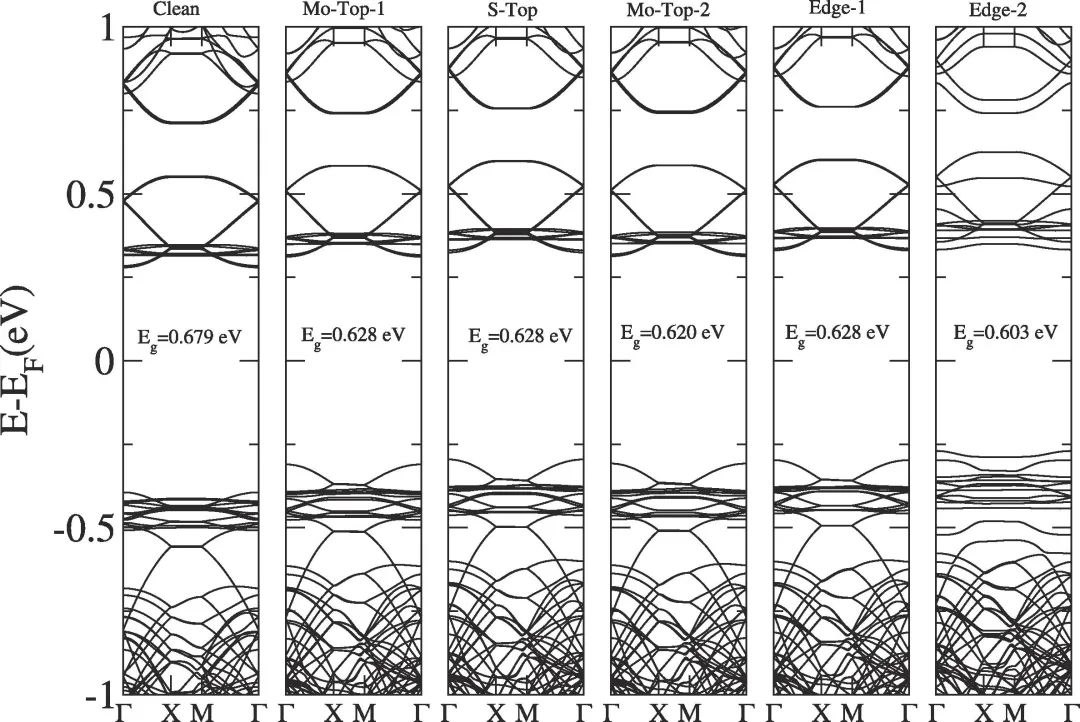
六、能带
能带计算可以分析MoS2材料的费米能级附近的能带分布,以及相应的带隙宽度,深度研究其导电性能和电催化性能。
华算科技已向国内外1000多家高校/科研单位提供了超过50000项理论计算和测试表征服务,部分计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!