

说明:本文华算科技系统介绍了杂化轨道基础理论,包括价键理论、杂化轨道理论、分子轨道理论和前沿轨道理论,并详细探讨了d-p、d-d、d-f及d-p-f等轨道杂化类型及其对催化性能的影响。读者可系统学习到轨道杂化的基本原理、分类及其在电催化中的作用机制,了解如何通过轨道工程优化催化剂设计,提升催化性能。
什么是杂化轨道?
杂化轨道理论由鲍林和斯莱特提出,是价键理论的延伸,用于解释价键理论无法描述的几何构型。该理论认为,原子轨道会混合形成适合化学成键的新杂化轨道。这些杂化轨道具有特定的空间取向,能在成键时最大化轨道重叠,从而形成更强、更稳定的化学键。
杂化轨道基础理论
价键理论(VB)
价键(VB)理论强调原子轨道的重叠以形成共价键。根据价键理论,当原子相互靠近时,分子系统的能量会降低,通过共享电子对形成共价键。利用价键理论可以分析反应中间体的成键状态,从而帮助我们评估反应物在催化剂表面的吸附能,并预测催化性能。


图1. (a)Fe 3d带中心与费米能级的相对位置对Fe-O键形成难易程度的影响(b)轴向氧配体示意图。DOI: 10.1016/j.cej.2024.155005
例如在氧还原反应(ORR)中,过渡金属基催化剂的未成对d轨道电子会与O₂的p轨道电子杂化并成键,从而激活吸附的O₂分子。不过,价键理论存在局限性,例如难以解释CH₄等分子中碳的四价性(碳会形成四个等效键,而非该理论单独预测的两个),且无法有效描述几何构型,这也促成了杂化轨道理论的发展。
杂化轨道理论(HO)
杂化轨道理论是价键理论的扩展,用于解释价键理论无法描述的几何构型。该理论认为,原子轨道会混合形成新的杂化轨道,这些杂化轨道在空间上具有特定取向,能在成键时最大化轨道重叠,从而形成更强、更稳定的化学键。
当所涉及的活性中心和反应物的轨道能级紧密排列并表现出更高的空间对称性时,产生的轨道杂化相互作用会更强。分析杂化轨道可以帮助研究人员理解反应路径、优化反应选择性并降低反应势垒。

图2. 反应表面与吸附物(Ads.)之间键形成的示意图。DOI: 10.1007/s40820-024-01528-9
例如,DFT计算表明,La的引入能优化NiFe基催化剂中d轨道与氧的2p轨道之间的杂化,增强氧中间体(*OOH)的吸附强度,降低OER决速步(*O→*OOH)的反应能垒,从而提升OER性能。
分子轨道理论(MO)
分子轨道(MO)理论与价键理论和杂化轨道理论不同,分子轨道理论将重点从单个原子相互作用转移到分子系统中电子的集体行为。当原子成键时,电子占据延伸到整个分子的分子轨道,共同参与化学键的形成。MO理论以原子轨道线性组合原理为基础,该原理将轨道分为三类:成键轨道、反键轨道和非键轨道。

图3. 通过原子沉积和原子植入方法对A层进行剥离以及过渡金属(TM)改性以制备h-MBenes的过程示意图。DOI: 10.1021/acsami.1c16449
在电催化反应中,分子轨道理论可用于描述催化剂和反应物之间的电子转移和轨道相互作用,将催化剂-反应物键的强度与反键轨道的占据联系起来。
例如,在六方MBene(Hf2BO2)基体掺杂后Mo金属的HER性能增强。掺杂原子的引入促进了O-2p轨道与H-1s轨道的成键,导致反键轨道的能量升高。这种相互作用增强了H─O键,从而提高了整体催化活性。
前线轨道理论(FO)
前沿轨道(FO)理论是分子轨道理论的具体应用,强调最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)在化学反应中的关键作用。HOMO由于其相对较低的结合能而起到电子供体的作用,LUMO则可以作为电子受体,从而促进催化过程中的电子转移。

图4. 通过DFT计算得到的能级图。DOI: 10.1016/j.cej.2023.144107
HOMO-LUMO间隙(Eg)可用于预测催化剂的整体反应性,Eg值越小越有利于化学反应。以HER为例,催化剂的HOMO和LUMO分别控制质子的吸附和还原能力。DFT计算表明,与纯Cu和MoO2相比,异质结构Cu/MoO2显示出最低的LUMO和Eg值,表明其更适合作为高效的HER催化剂。
轨道杂化的分类
根据涉及的轨道数量,轨道杂化可分为两类:(1)二元轨道杂化,包括常见的杂化如s-s、s-p、s-d、p-p、d-p、d-d和d-f。(2)多轨道杂化,包括更复杂的构型如d-p-f或级联轨道杂化。
d-p轨道杂化是一种常见的二元轨道杂化类型,主要涉及过渡金属的d轨道与p区元素(如S、O、N、P等)的p轨道之间的相互作用,通过电子耦合影响催化过程。其在电催化中发挥重要作用:可增强催化剂与中间体的结合亲和力,优化电子结构,促进反应物活化与转化,提升催化活性、选择性和稳定性。

图5.(a)Sn-Ru/C中Ru和Sn位点之间不同轨道相互作用的示意图。(b)Sn-Ru中Ru的d轨道和Sn的p轨道、Ga-Ru中Ru的d轨道和Ga的p轨道以及Ru中Ru的d轨道的部分态密度(PDOS)。d带中心以虚线标记,基于费米能级(费米能级对齐在0 eV)。DOI: 10.1021/acscatal.2c05547
例如通过将Sn/Ga杂元素掺杂到Ru主体中,开发了一种d-p轨道杂化催化剂。PDOS图分析表明,这种d-p杂化改变了Ru的电子结构,从而提高了氢氧化反应(HOR)的催化活性。
d-d轨道杂化的特征是过渡金属原子之间不同d轨道的相互作用和混合。这种杂化导致d轨道分裂为不同的能级,这对催化剂的电子性质有深远影响。在许多电催化系统中,d-d杂化可以决定未成对电子的可用性以及催化剂给出或接受电子的能力。

图6. M-WO₂(M = Fe、Co、Ni、Cu等)中W 5d-M 3d轨道调制的示意图。DOI: 10.1002/aenm.202103301
有研究使用WO₂作为HER的模型催化剂,进一步探索了d-d轨道耦合与催化性能之间的相关性。通过引入各种过渡金属杂原子来修饰d轨道杂化,研究人员发现用Fe、Co、Ni或Cu取代W位点形成W─M键会增加d轨道的占据率,导致这些W─M键内反键轨道的填充增强。
理论计算表明,在WO₂晶格中引入金属杂原子通过调控W 5d-M 3d轨道杂化,与原始的W─W构型相比,有效减弱了W─M位点的氢吸附。
d-f轨道杂化指过渡金属的d轨道与稀土金属(如Ce、La、Nd、Sm等)的f轨道之间的相互作用,这类杂化因稀土金属独特的价电子构型(如4fn-15d16s2或4fn6s2)及5s、5p电子的屏蔽效应而具有特殊性。
首先,d-f杂化可以显著改变关键中间体(如氢和氧物种)的吸附能。通过优化反应路径和降低能垒,d-f杂化最终提高了催化剂体系的整体电催化活性和稳定性。

图7. (e)在NFC和NFC-CeO₂上进行OER的能量路径。(f)NFC-CeO₂通过d-f电子梯提升OER的示意图。DOI: 10.1002/adfm.201908367
其次,d-f轨道杂化有可能加速电子转移。引入CeO₂作为镍-铁-铬氢氧化物(NFC)的载体,已被证明能提高OER性能,在碱性溶液中实现230.8 mV的超低过电势(10mA cm-2)和32.7mV dec-1的塔菲尔斜率,及优异的耐久性。
DFT计算表明,通过NFC和CeO₂之间的相互作用形成的d-f电子阶梯显著促进了高速电子转移。
第三,d-f轨道杂化可以诱导活性中心的重构,这种现象通常发生在OER过程中。这种重构增强了活性中心对含氧化中间体的识别能力,从而提高了催化活性。例如,通过引入CeOx开发了一种基于高密度CoOx催化剂的新型双功能电催化剂,其中Ce通过破坏表面Co物种的电子结构促进CoOOH的有效形成。
d-p-f轨道杂化属于多轨道杂化,涉及过渡金属的d轨道、p区元素的p轨道(如O的2p轨道)与稀土金属的f轨道(如Ce的4f、Eu的4f轨道)之间的相互作用,通过提供更丰富的轨道组合和能级,优化电催化过程中的反应物吸附、活化及反应路径,提升催化活性、选择性和稳定性。
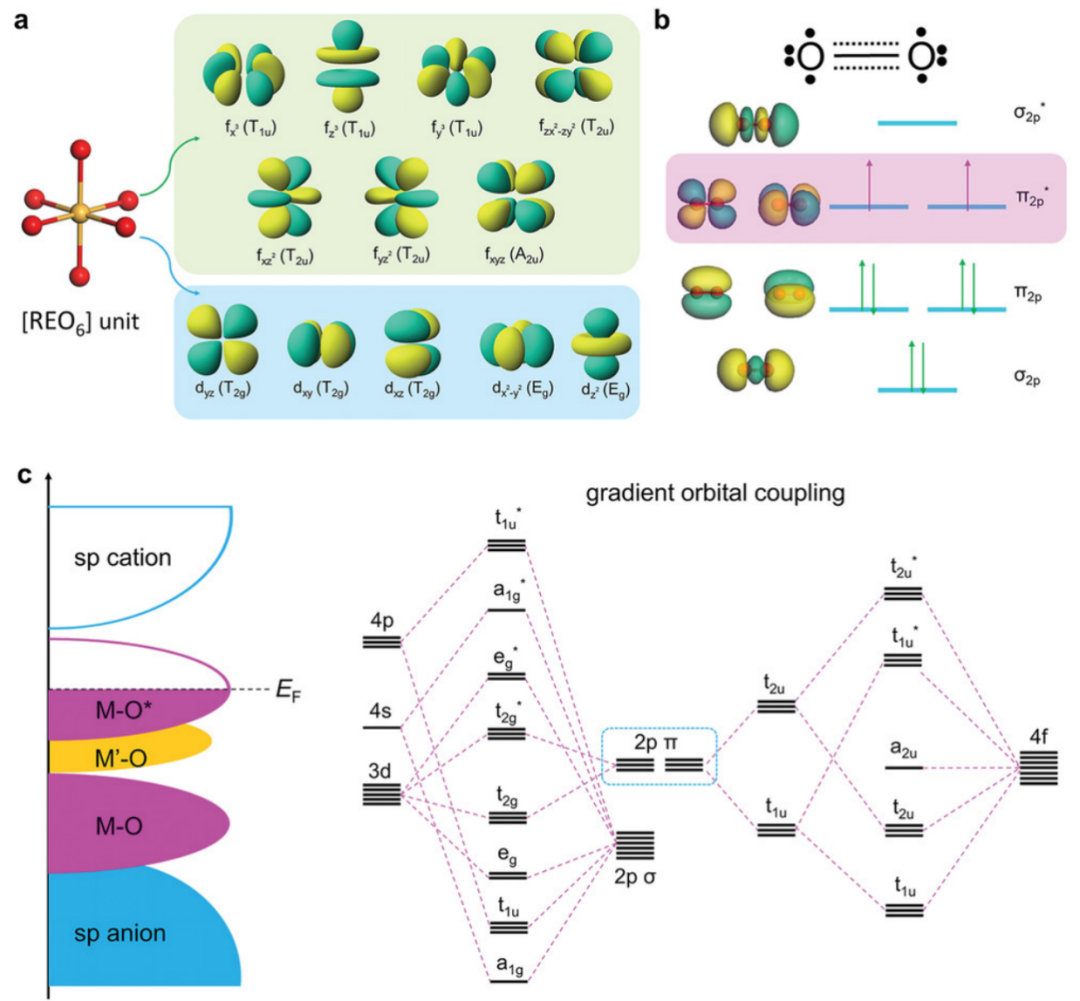
图8.(a)基于轨道耦合工程的稀土(RE)基材料合理设计的理论预测。(b)O2的分子轨道能级图。(c)通过梯度3d-2p-4f轨道耦合的金属-氧-金属相互作用的示意图,并附有定性分子轨道图。DOI: 10.1002/adma.202206540
其作用机制主要包括:稀土金属的f轨道因能级较高且靠近费米能级,可作为电子补偿器或泵,通过金属-氧-金属相互作用促进电子离域,激活惰性电子态;同时,梯度轨道耦合(如(Co)3d-(O)2p-(Eu)4f)能优化活性中心的电子结构,平衡电子转移与中间体吸附。
【做计算 找华算】
华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。