

说明:本文华算科技系统介绍了氧化铈(CeO₂)在计算化学中的关键研究方向:通过DFT+U/混合泛函解析氧空位与Ce 4f电子的本质,计算吸附与反应能垒揭示催化机理,利用AIMD/迁移能计算评估氧离子扩散与动力学行为,并通过界面与掺杂模拟为材料改性提供设计思路。文中强调模型选择与参数校准对结果可靠性的影响。
CeO₂(氧化铈)是一种在催化、传感和能源材料里经常出现的“多面手”。它最有名的特性是“可储氧/释氧”(oxygen storage capacity),也就是说它可以通过形成和修复氧空位在氧化还原循环中发挥缓冲作用。
下图展示了分别从侧面和顶部视角展示了完美CeO₂(111)表面和还原后含氧空位(VO)的CeO₂(111)表面的原子结构,标注了Ce³⁺、Ce⁴⁺和氧空位的位置。
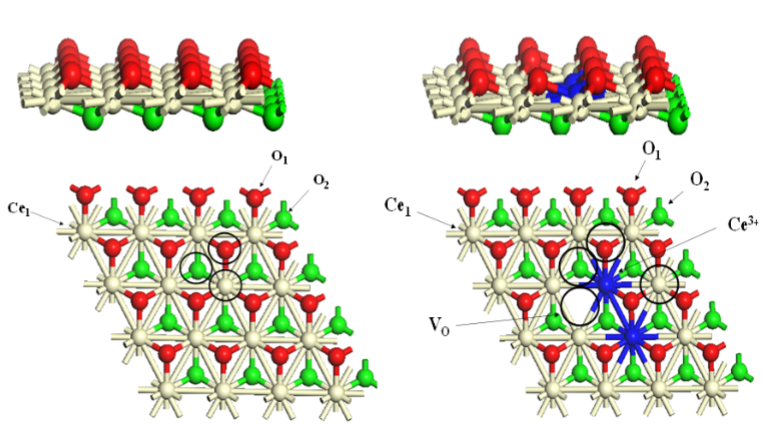

DOI:10.3390/molecules26216485
对于初学者来说,想象CeO₂像一个可以暂时藏氧的小仓库:当环境还原时仓库放出氧(形成空位),当环境氧化时仓库又把氧收回来。这样的可逆性让CeO₂在三元催化剂、固体氧化物燃料电池、电催化和污染治理中非常关键。
计算化学能帮我们从原子尺度理解这些“藏氧”过程:哪里更容易形成空位,空位如何影响电子结构,空位附近的吸附分子怎样被活化,这些都是计算可以量化并预测的问题。
在理论模拟里,CeO₂的一个挑战是钬(f)电子和氧空位导致的强局域化效应,因此常用的工具是DFT+U或混合泛函来更准确地描述Ce 4f轨道。通过这些方法可以计算氧空位形成能、空位的稳定位点、以及空位对局域电子(小极化子)和费米能级的影响。
计算还能预测吸附能和反应路径:把CO、H₂O或NO等分子放到CeO₂表面,用DFT和过渡态搜索(如NEB)找到其吸附构型和反应能垒,从而解释催化选择性和活性差异。
下图展示了Hg⁰在CeO₂(111)表面的四种稳定吸附构型(标记为1A、1B、1C和1D),包括顶视图和侧视图。这些构型显示了Hg原子与表面Ce或O原子的距离。
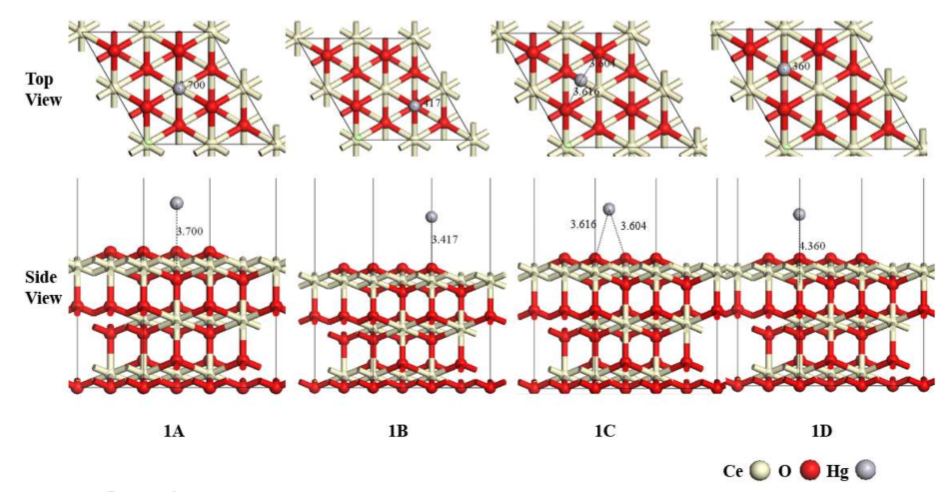
DOI:10.3390/ma11040485
此外,电子态密度(DOS)、Bader电荷分析和电荷密度差映射能直观显示氧空位或掺杂如何改变局域电荷分布,这对理解CeO₂的还原性和与金属颗粒的相互作用非常重要。
静态能量计算只能告诉我们“能否发生”,而动力学模拟(AIMD或经典MD配合机器学习势)能展示温度下空位迁移、氧扩散和吸附物的动态演化。计算氧离子在晶格或表面间迁移的迁移能垒以及扩散系数,有助于评价材料在实际器件中的响应速度。
界面模拟也很重要:CeO₂常作为载体与金属(如Pt、Pd)形成复合催化剂,界面处的电子转移与氧空位生成常常主导催化性能,QM/MM或嵌入式DFT能兼顾界面化学与尺度问题。
从材料设计角度看,计算提供了掺杂策略(用稀土或过渡金属改变空位形成能)、表面工程(不同晶面与缺陷密度)和高通量筛选的思路,帮助把原子级理解转化为可合成的改性建议。
DFT计算模型显示了CeO₂/Co₃O₄界面的原子结构,其中氧空位优先在界面附近形成。电子密度差图显示电子从CeO₂向Co₃O₄转移,导致Co原子电子密度增加,同时Ce³⁺浓度升高,促进氧空位生成。
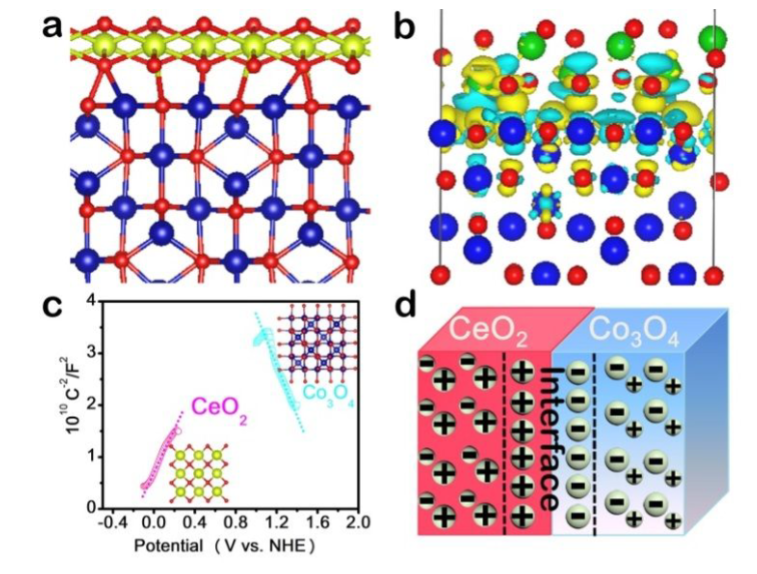
DOI:10.1021/acscatal.9b01819
实务上建议初学者关注U值与泛函敏感性、尺寸与真空层的收敛、带自旋极化的计算,以及对有电荷态缺陷的有限尺寸修正和自由能(含振动项)评估,结合光谱(XPS、Raman)或热化学数据校准模型,能显著提高计算的可用性和可靠性。
CeO₂以其可逆的氧化还原能力和丰富的表面化学成为催化与能源领域的重要材料。
计算化学可从电子结构、缺陷热力学、吸附反应路径到动力学扩散全方位描述其行为:DFT+U或混合泛函适合处理Ce 4f电子与氧空位,过渡态搜索揭示化学反应的能垒,AIMD与迁移路径计算提供温度依赖和时间尺度的信息,界面模拟则还原与金属或电解质相互作用的真实情景。
实际研究中应注意泛函与U参数的选择、有限尺寸与电荷修正、振动自由能的贡献,以及与实验光谱和热化学数据的比对。结合高通量筛选和机器学习势,计算能显著加速CeO₂相关催化剂与功能材料的设计,使理论预测更快地指导实验验证与工程应用。
【做计算 找华算】
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???