

说明:分子模拟技术被誉为“计算显微镜”,它使我们能在原子尺度上观察和理解物质的行为。而势函数(或称力场)正是这台显微镜的“物理定律”核心。
本文华算科技旨在系统阐述势函数的定义、其在驱动分子模拟中的核心作用,并梳理当前主流的力场类型,以揭示其在现代科学研究中的重要性。



什么是势函数


势函数(Potential Function),在分子模拟领域通常被称为力场(Force Field),是一套用于描述分子体系势能(V)与其三维结构(即所有原子的坐标 r)之间关系的数学经验函数 (经典分子模拟理论)。
其核心思想是将一个复杂多体系统的总势能分解为一系列相对简单的、可计算的能量项的总和。这个函数构成了所谓的势能面(Potential Energy Surface, PES),是决定分子构象、动力学行为和热力学性质的理论基础。
DOI: 10.1038/s41524-024-01272-z
一个典型的经典势函数通常包含两大类相互作用:
成键相互作用(Bonded Interactions):这些是作用于通过共价键直接或间接连接的原子之间的“局部”相互作用,用来维持分子的基本骨架。
键长伸缩(Bond Stretching):通常用简谐振子模型(如胡克定律)来描述,能量与键长偏离其平衡值(r0)的平方成正比:Vbond=kb(r−r0)2。这反映了拉伸或压缩共价键所需的能量。
键角弯曲(Angle Bending):类似地,原子间形成的键角也会在平衡角度(θ0)附近振动,其能量贡献通常也用谐振势描述:Vangle=kθ(θ−θ0)2
二面角扭转(Dihedral Torsion):描述了围绕一根化学键旋转时能量的变化,这对于确定分子(尤其是蛋白质、聚合物等柔性大分子)的构象至关重要。它通常由一个周期性的余弦函数来表示:Vdihedral=∑Vn[1+cos(nφ−γ)]。
非键相互作用(Non-bonded Interactions):这些是作用于所有原子对(通常排除通过少数几个共价键直接相连的原子对)之间的“远程”相互作用,决定了分子的空间堆积、折叠和分子间识别。
范德华相互作用(van der Waals Interaction):它同时包含了短程的排斥力(源于泡利不相容原理)和长程的吸引力(源于瞬时偶极诱导的色散力)。最经典的数学形式是Lennard-Jones (12-6)势:VLJ=4ε[(rσ)12−(rσ)6]。其中,r12项主导排斥,r6项主导吸引。
静电相互作用(Electrostatic Interaction):基于库仑定律,描述了原子部分电荷(qi, qⱼ)之间的相互作用:Vcoulomb=(qiqj)/4πε0εrrij。这是驱动极性分子相互作用、离子识别和氢键形成的关键力量。
因此,系统的总势能可以写为这些能量项的总和:
Vtotal=Vbond+Vangle+Vdihedral+VLJ+Vcoulomb
这些公式中的参数共同构成了“力场参数集”。这些参数是通过拟合大量的量子化学计算结果和实验数据(如晶体结构、振动光谱、热力学数据)得到的,其准确性直接决定了模拟结果的可靠性。
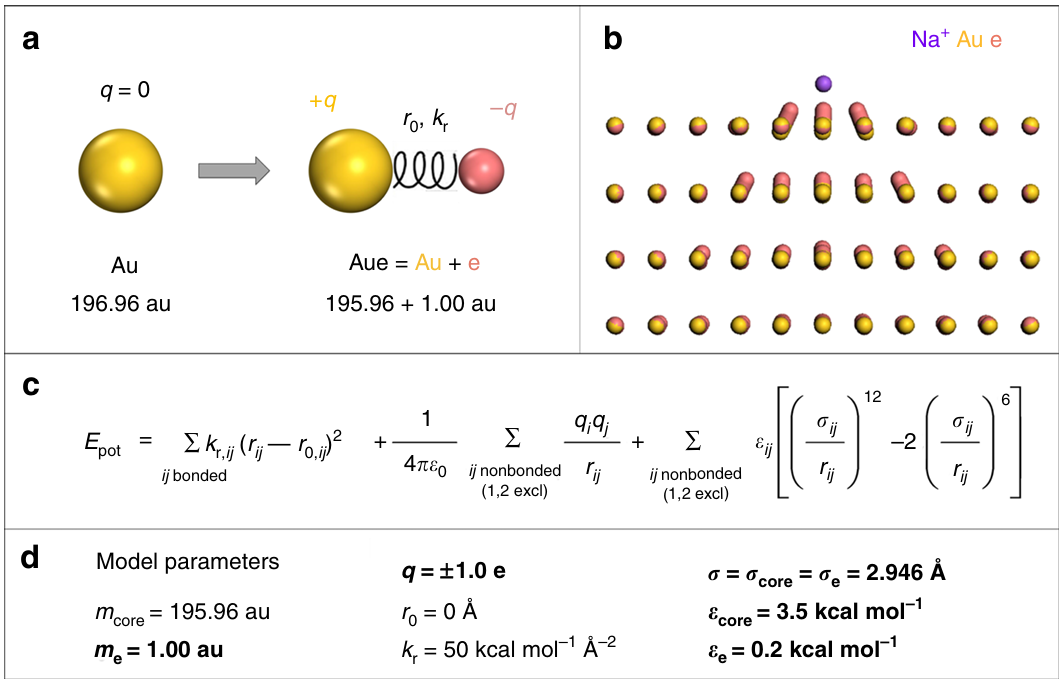
DOI: 10.1038/s41467-018-03137-8
势函数的应用
势函数是分子动力学(Molecular Dynamics, MD)模拟的引擎。模拟过程本质上是在势能面上求解牛顿第二运动定律(F = ma)。其具体步骤如下:
计算力:在模拟的任意一个时间点,体系中每个原子所受的合力(F)可以通过对总势能函数(V)求其位置坐标的负梯度来得到:Fi=−∇iVtotal。这一步是整个模拟中计算量最大的环节,因为它需要计算体系中所有原子对之间的非键相互作用。
更新运动状态:根据计算出的力F和原子的质量m,可以得到该原子的加速度a(a = F/m)。
时间积分:使用一种数值积分算法(如Verlet或Leap-frog算法),在一个极小的时间步长(Δt,通常为飞秒级别)内,根据当前的位置、速度和加速度,来预测原子在下一个时刻(t+Δt)的新位置和新速度。
迭代循环:重复上述“计算力->更新运动状态”的循环,模拟就可以一步步地在时间维度上推进。这个过程最终会生成一个原子坐标随时间变化的轨迹文件,就像一部“分子电影”。
通过分析这条轨迹,研究者可以获得丰富的动态和静态信息,例如观察蛋白质如何折叠、药物分子如何与靶点结合、材料的力学性质如何响应外界刺激,以及计算系统的自由能、扩散系数等宏观热力学和动力学性质。可以说,势函数将静态的结构信息转化为了动态的物理过程。
常见的力场类型
全原子力场(All-Atom Force Fields):这是最精细的力场之一,明确地处理系统中的每一个原子,包括氢原子。它们能够提供非常详细的相互作用信息,特别适用于需要精确描述氢键和静电相互作用的体系,如蛋白质、核酸和水的模拟。代表性的力场有AMBER, CHARMM, OPLS-AA等。
联合原子力场(United-Atom Force Fields):为了提升计算效率,这类力场将非极性的氢原子(如脂肪链上的-CH2–和-CH3基团)与其所连接的重原子(如碳原子)合并成一个“联合原子”作用中心。这显著减少了系统中的粒子总数和需要计算的相互作用项,从而加快了模拟速度。GROMOS力场是其典型代表,常用于脂质和聚合物的模拟。
粗粒化力场(Coarse-Grained, CG Force Fields):当研究尺度非常大(如细胞膜、病毒衣壳)或时间尺度非常长(如毫秒到秒级别)时,原子级别的模拟变得不切实际。粗粒化模型将一组原子(如一个氨基酸残基、一个核苷酸或几个水分子)简化为一个“珠子”(bead)。这极大地减少了系统的自由度,使得模拟更大、更慢的生物过程成为可能,但代价是牺牲了原子级别的细节。著名的例子是MARTINI力场。
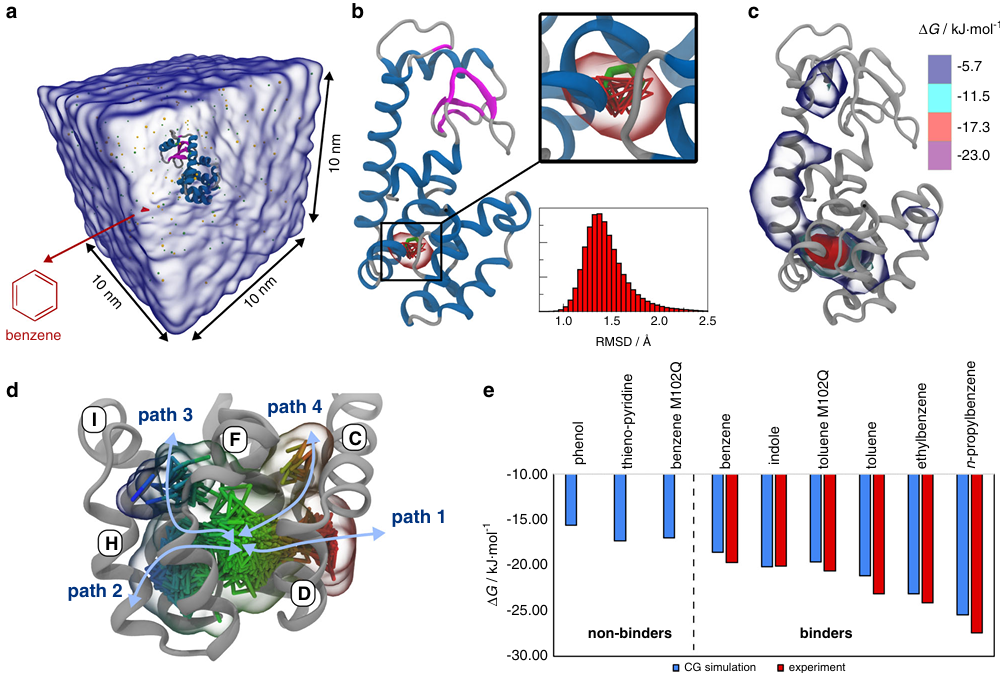
DOI: 10.1038/s41467-020-17437-5
可极化力场(Polarizable Force Fields):传统的力场使用固定的原子点电荷,这无法描述分子因环境电场变化而产生的电荷重新分布(即极化效应)。可极化力场通过引入浮动电荷、偶极子或Drude振子等模型,动态地调整原子的电荷分布,从而更精确地描述离子–水相互作用、金属离子催化等对静电环境高度敏感的体系。例如AMBER-f/f, CHARMM-Drude和AMOEBA。这类力场精度更高,但计算成本也急剧增加。
反应力场(Reactive Force Fields):经典力场无法描述化学键的生成与断裂。为了模拟化学反应过程,反应力场(如ReaxFF)被开发出来。它使用与键级(bond order)相关的复杂函数形式来描述原子间的相互作用,使得共价键可以在模拟过程中动态地形成和断开,从而架起了经典分子动力学与量子化学计算之间的桥梁。
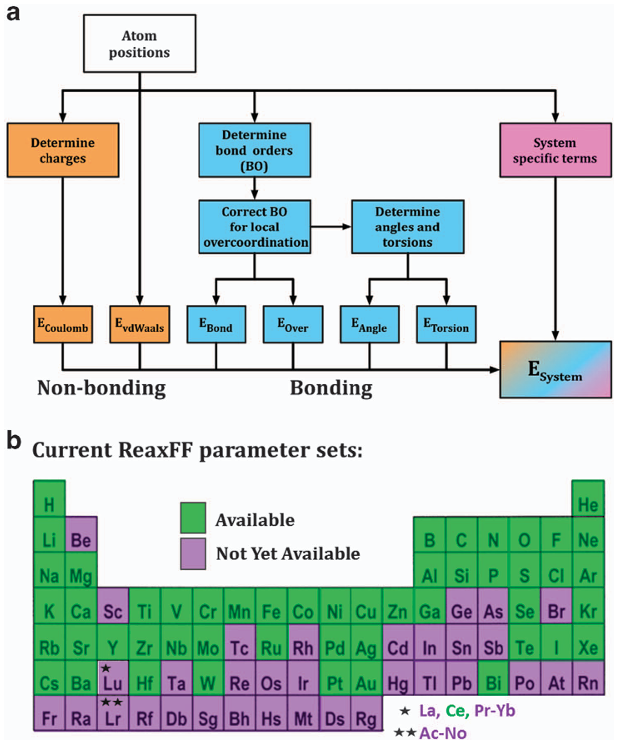
DOI: 10.1038/npjcompumats.2015.11
小结
势函数是经典分子模拟的理论灵魂,它以经验性的数学公式近似地描绘了原子间的相互作用势能面。通过在这势能面上求解牛顿运动方程,我们得以窥见微观世界的动态景象。
从高精度的全原子力场到大尺度的粗粒化力场,不同类型的势函数体现了在模拟精度与计算效率之间的持续权衡与选择,为解决不同层次的科学问题提供了强有力的理论工具。
【做计算 找华算】
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???