

说明:本文从理论计算的角度,系统介绍催化活性位点(Catalytic Active Sites)的基本概念、核心原理及其在化学和生物催化中的研究进展。
内容涵盖催化活性位点的定义、热力学特性、计算方法(如密度泛函理论、分子动力学和机器学习)以及在酶工程、异相催化和药物设计中的重要性。
读者可通过本文了解催化活性位点的独特机制、模拟技术的关键作用,以及其在先进催化系统设计中的潜力,为计算化学、材料科学和生物工程的创新研究提供理论支持和实践指导。



什么是催化活性位点?
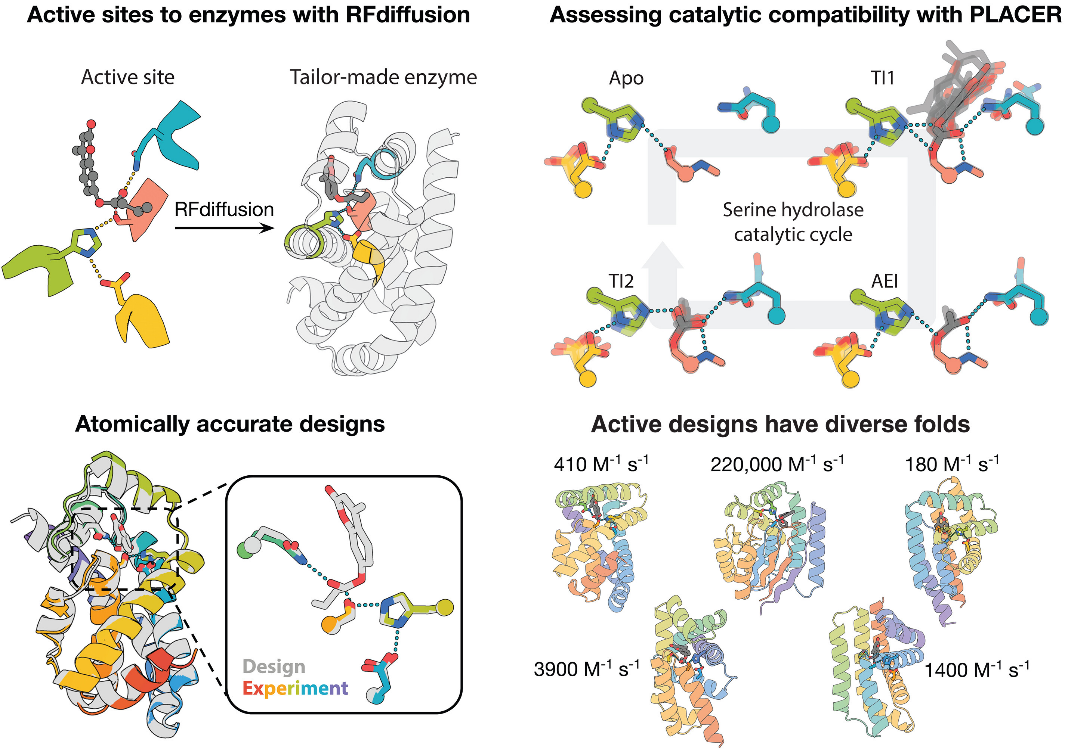
DOI: 10.1126/science.adu2454
催化活性位点是指催化剂或酶分子中直接参与化学反应、降低反应活化能的特定区域或原子簇。这些位点通常由氨基酸残基、金属离子或表面原子组成,通过与底物形成过渡态,实现高效的催化过程。
在酶催化中,活性位点往往位于蛋白质的腔体中,提供精确的立体环境和化学功能;在异相催化中,活性位点可能为纳米颗粒的边缘、台阶或缺陷位。催化活性位点的核心原理源于过渡态理论,其中位点稳定中间体,降低反应势垒,从而加速反应速率。
传统实验方法如X射线晶体学可表征位点结构,但理论计算方法在揭示动态行为和电子转移机制方面具有独特优势。
催化活性位点的理论计算方法
理论计算在催化活性位点研究中扮演关键角色,用于预测位点结构、反应机制和性能优化。以下介绍主要计算方法及其在活性位点中的应用。
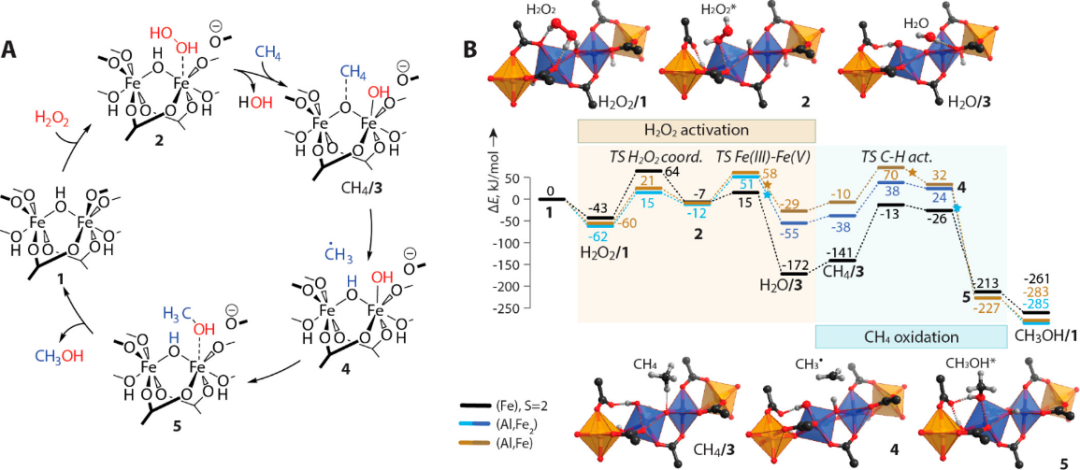
DOI: 10.1021/acs.chemrev.8b00361
密度泛函理论基于量子力学,计算活性位点的电子结构、结合能和反应势垒。其核心优势是无需经验参数,直接从电子层面预测原子间相互作用和催化活性。
例如,嵌入八面体结构形成AlOx链中的单位和双位O型桥接铁配合物被发现能在氧化性H2O2环境中足够稳定,同时还能形成低能量反应路径,从而实现甲烷的选择性氧化为甲醇。
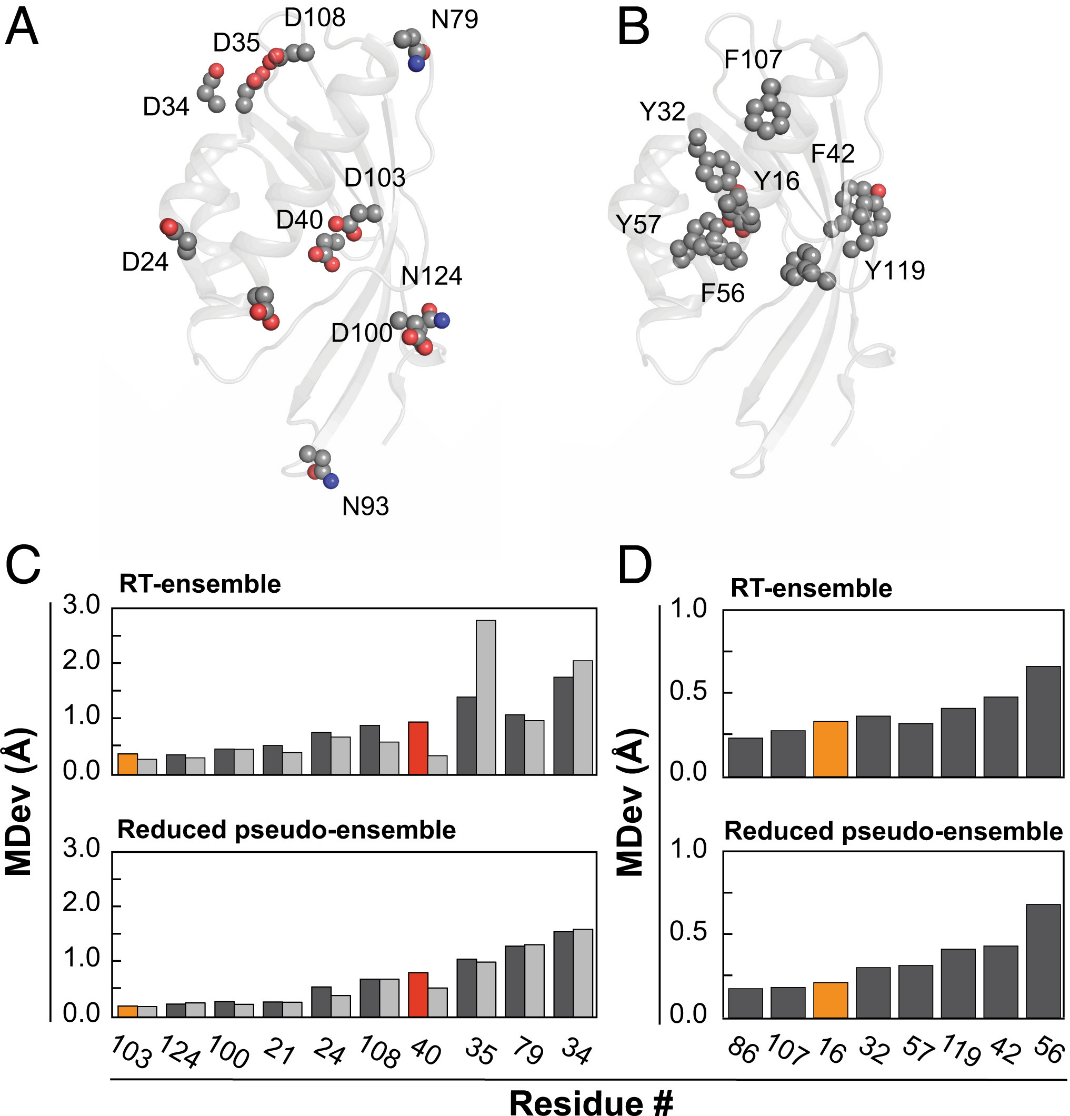
DOI: 10.1073/pnas.2011350117
分子动力学通过牛顿力学模拟原子运动,研究活性位点的动态行为,如底物扩散和构象变化。MD依赖于力场模型,适合大尺度体系模拟。
在催化活性位点中,MD常用于分析酶的柔性循环对位点访问的影响,以及溶剂效应。例如,MD模拟揭示了酮甾异构酶(KSI)活性位点的ensemble行为对催化效率的贡献。
MD在活性位点工程中应用广泛,如预测突变对酶活性的影响和异相催化中的表面重构。挑战在于力场准确性,需通过QM/MM混合方法提升精度,结合实验数据校正。
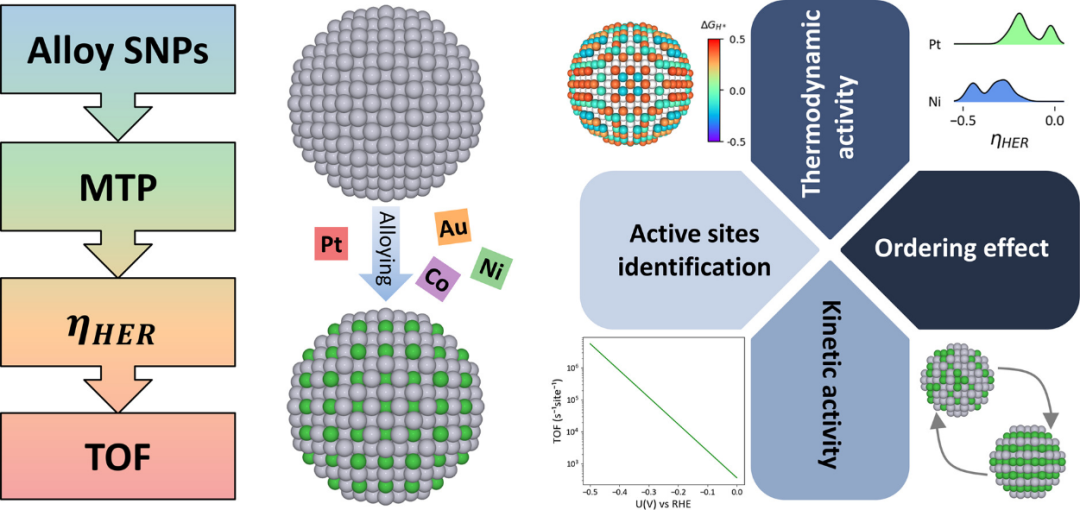
DOI: 10.1016/j.cej.2025.167929
机器学习通过数据驱动方法,优化活性位点的计算效率和精度。例如,基于机器学习的框架,通过将矩张量势(MTP)与主动学习相结合来预测合金球形纳米颗粒(SNP)的性能。
这种方法能够有效评估各种形态、成分和粒径的结合自由能和周转频率 (TOF),包括PtAu(核壳)、PtCo(尺寸/成分依赖性)和PtNi(固溶体/金属间化合物)。催化活性评估揭示了实现最高HER性能的最佳成分。
结论
催化活性位点作为催化反应的核心,通过精确的结构和动态机制实现高效转化,成为化学和生物工程的焦点。理论计算方法——密度泛函理论、分子动力学和机器学习——为活性位点的机制解析和优化提供了强大工具。
这些方法通过多尺度建模和数据驱动策略,显著推进了酶工程、异相催化和能源转换中的应用。随着计算技术和算法的进步,催化活性位点的设计将进一步加速,为绿色化学和可持续技术提供新机遇。
【做计算 找华算】
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???