

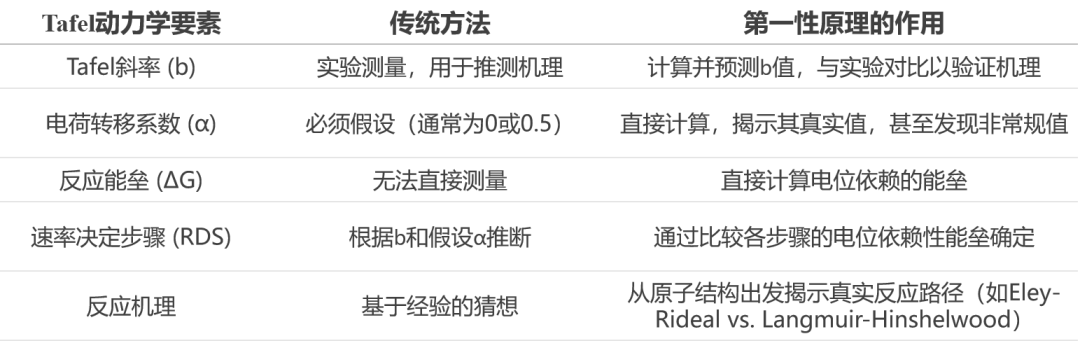
说明:本文华算科技系统解析“塔菲尔动力学”实验规律与“第一性原理计算”的结合方式。随着第一性原理计算技术的发展,尤其是DFT/CM-MPB等方法的应用,研究人员能够在原子尺度上理解电位对反应动力学的影响,预测Tafel斜率,并揭示反应机理。


塔菲尔斜率的理论基础

电化学反应的动力学研究一直是理解与设计高效电催化剂的核心。Tafel方程由瑞士化学家Julius Tafel于1905年提出,建立了电流密度i与过电位η之间的对数关系:
η=a+blogi
其中,b为Tafel斜率,与电荷转移系数(CTC,α)相关:
b=2.3RT/αF
Tafel方程不仅适用于氢析出反应(HER),还广泛应用于氧析出(OER)、氧还原(ORR)、甲醇氧化(MOR)等反应,是分析电极反应机理和速率决定步骤(rds)的重要工具。
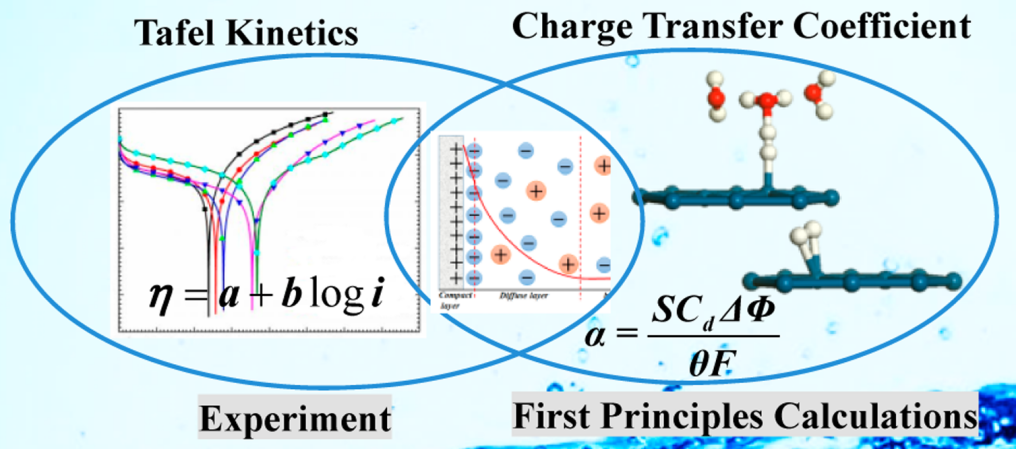
Tafel最初在研究有机物的电还原反应时发现,HER是主要的竞争反应。通过大量实验数据总结出电流与电位之间的对数关系,即Tafel方程。
后来,Volmer和Buller等人将Tafel方程与Arrhenius方程和过渡态理论结合,推导出电化学反应速率与电位的关系:
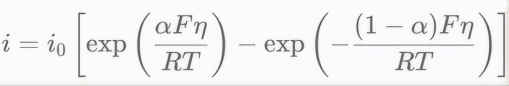
在高过电位下,可简化为Tafel形式。Tafel斜率b和截距a都与α相关反映了电子转移的本质。
Tafel斜率的实验观察与机理分析
Tafel斜率的变化常反映反应机理的转变。例如,OER在RuO2上在低过电位下斜率为59 mV,高过电位下为118 mV,表明Tafel斜率可能随电位变化。
表1. 重要电催化反应的实测Tafel斜率
第一性原理计算的物理模型
双电层模型:该图清晰地表明了电化学界面不是一个简单的、理想的二维平面,而是一个具有复杂三维结构的区域。
它被分为两个主要部分:紧密层(Stern or Helmholtz Layer):最靠近电极的一层,包含特异性吸附的离子或分子(如水分子),其结构主要由原子间的短程相互作用(化学键、范德华力)决定。
扩散层(Diffuse Layer):紧挨着紧密层,向外延伸至本体溶液。包含非特异性吸附的离子,其分布由静电作用和热运动(熵效应)共同决定,遵循Poisson-Boltzmann分布。
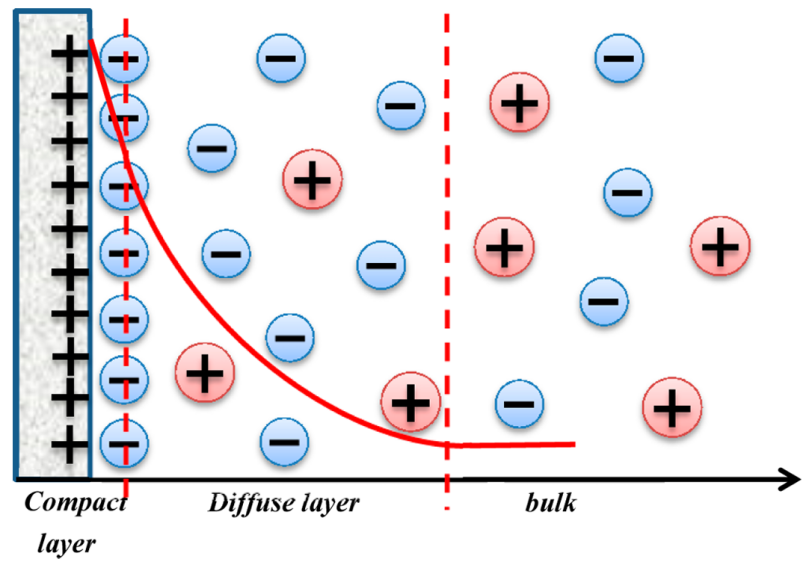
图1. Gouy-Chapman-Stern双电层模型。DOI: 10.1021/cs501312v。
任何试图模拟电化学体系的理论方法,都必须能够描述或重现这种结构。
第一性原理计算的目标就是在原子尺度上“看到”并理解这个结构。
它定义了理论计算需要模拟的物理对象,解释了“电位”的微观含义,并直接启发了文中核心计算方法(DFT/CM-MPB)的架构设计。
Tafel斜率动力学与第一性原理计算
宏观实验现象:实验上测量得到一条电流密度(j)对过电位(η)的曲线,即Tafel曲线。通过分析其斜率(Tafel斜率,b)和截距(与交换电流密度j0相关),电化学家可以对反应机理(如速率决定步骤RDS)进行推测。
推测的局限性:这种推测依赖于预先对基元步骤的电荷转移系数(α)做出假设(通常假设为0或0.5)。然而,对于复杂的表面反应,α可能是一个非常规值,导致基于传统假设的机理分析失效或产生争议。
第一性原理计算(微观):密度泛函理论(DFT)等第一性原理方法的作用就是直接计算这些原本需要假设的关键微观物理量。
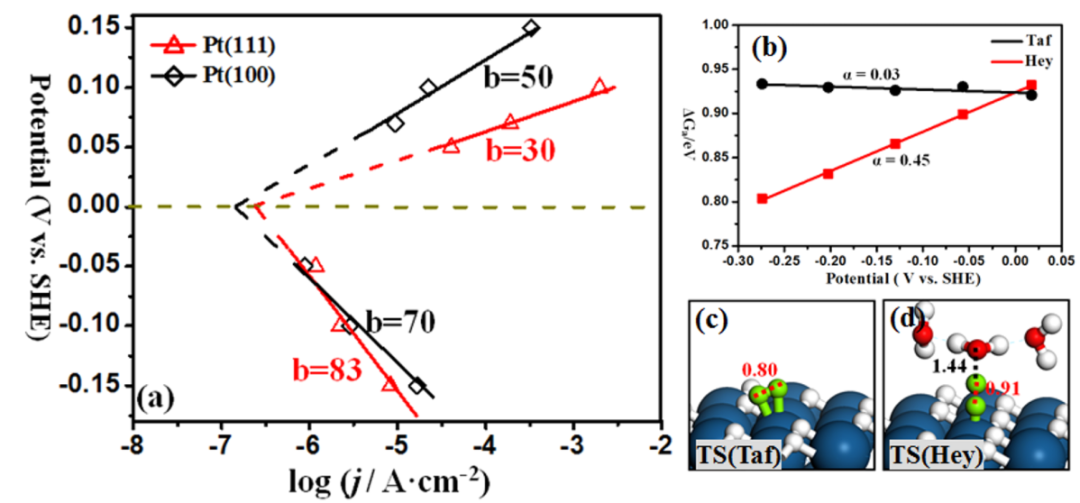
图2. 基于DFT/CM-MPB方法得到的HER/HOR塔菲尔动力学结果。DOI: 10.1021/jp400608p。
计算对象:在明确的电极电位(U)下,计算反应路径上各态(初始态IS、过渡态TS、最终态FS)的自由能(G)。
关键输出:反应能垒(ΔG):决定反应速率。
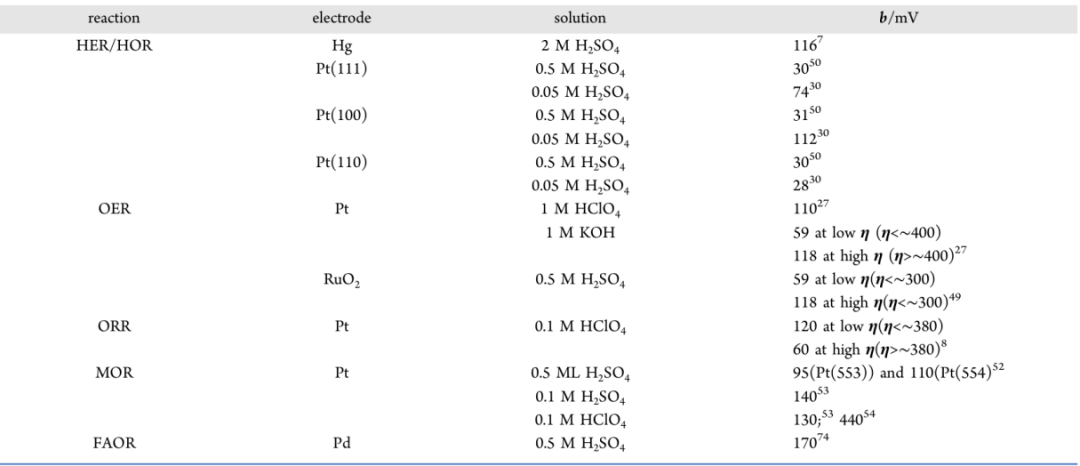
电荷转移系数(α):不再假设,而是通过计算能垒对电位的导数得到,这是连接微观计算与宏观Tafel斜率的最关键物理量。
机理验证与预测(宏观):将计算得到的真实α值代入微观动力学模型,可以从第一性原理直接推导出整个反应的Tafel曲线,从而与实验测量进行直接、定量的对比。计算得出的Tafel斜率b成为了验证理论模型正确性的最终判据。
理论模型与第一性原理计算方法
核心思想:该方法由Anderson等人于1999年提出。其关键是将电极电位与团簇模型的电子亲和能(EA)或电离能(IP)相关联。通过计算反应物的电离能(IP)和电子亲和能(EA)来估计反应电位。
方法详解:标准氢电极(SHE)的功势通常取为~4.6 eV。因此,一个反应在电位U(相对于SHE)下发生还原反应时,满足:EA=(4.6+U)eV。
通过量子化学计算,沿着反应坐标优化反应中心团簇(例如,对于HER,可能是Pt-H—H3O+(H2O)n)的结构,可以找到EA或IP等于(4.6+U)eV的点,该点即为该电位下的过渡态或反应发生点。
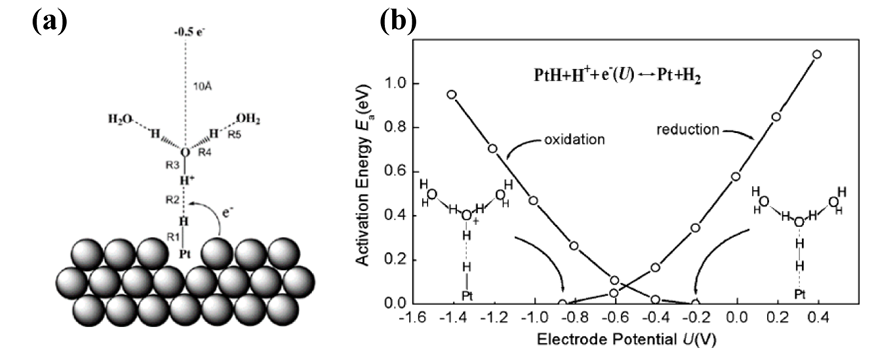
图3. 模型结构示意图及Pt上HER的能垒随电位的变化。DOI: 10.1021/jp037126d。
优点:概念清晰,计算量相对较小,能够提供电位依赖的能垒信息。
局限性:团簇模型过于简化,无法准确描述真实的周期性电极表面、表面电子结构(如d带中心)、吸附质覆盖度效应以及表面的长程电场效应。
核心思想:使用周期性平板模型来更真实地模拟电极表面,并通过向体系直接添加或移除电子来模拟充电效应。为了控制计算中的静电发散,需要一套严谨的方法来定义和计算绝对电极电位。
方法详解:该方法由Neurock等人发展。构建一个包含金属板层、真空层和数层显性水分子的超胞。水分子通常初始化为冰状结构以节省计算资源。
第一参考:计算中性体系时,在真空层中央插入一个测试质子,计算该点的静电势。第二参考:假设在充电时,水层中心的静电势保持不变。
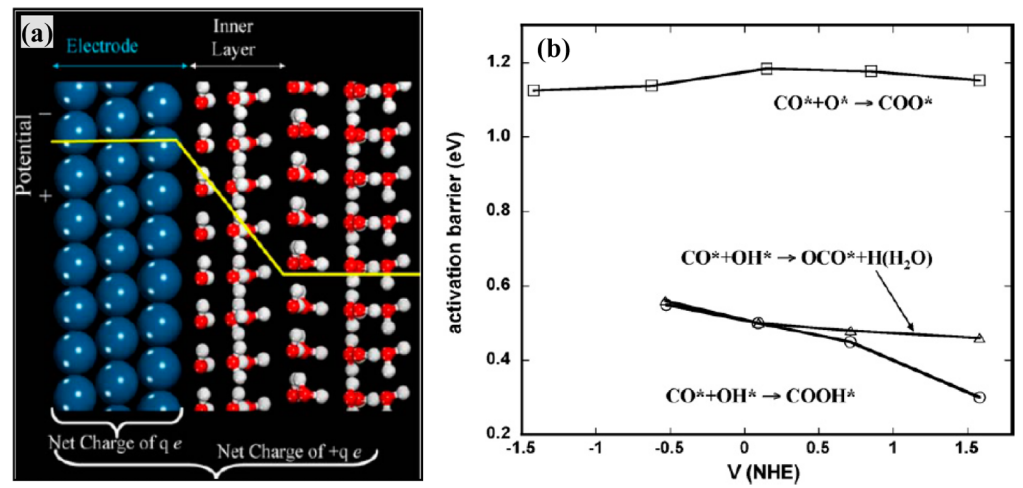
图4. 双参考方法示意图及CO氧化反应能垒与电位关系。DOI: 10.1007/s11244-007-9004-9;10.1016/j.electacta.2007.01.060。
优点:采用了更真实的周期性表面模型,可以考虑表面结构、覆盖度等效应。
局限性:使用静态的冰水结构无法模拟常温下水分子的动态行为和复杂的氢键网络。电位通过充电实现,但显性水层的结构可能随电位发生剧烈变化,这一点在静态计算中难以捕捉。
核心思想:采用隐式溶剂模型来替代大部分的显性水分子,从而极大地降低计算成本,同时又能捕捉到溶剂化带来的长程静电效应。该方法可以与带电的周期性平板计算自然结合。
方法详解:该方法将修改的Poisson-Boltzmann方程(MPB)与DFT计算耦合,自洽地求解电极/电解液界面的静电势。
该模型通过修改的Poisson-Boltzmann方程(MPB)模拟电解质中的长程静电作用,能自洽地处理表面电荷与溶剂化效应(图3)。
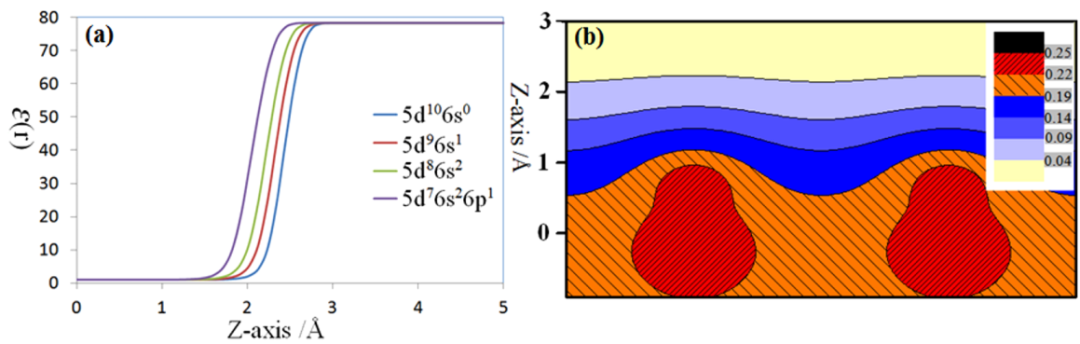
图5. Pt(111)表面的介电函数分布及溶剂化对静电势的影响。DOI: 10.1016/j.cattod.2012.04.055。
优点:计算高效,无需昂贵的显性水分子MD模拟;物理清晰,能很好地处理长程静电和电解质效应,自动给出双电层结构;适用性广,可方便地用于计算各种电位下的反应能垒、表面相图、微分电容等。
局限性:短程相互作用,对于需要与特定水分子形成短程强相互作用(如氢键)的反应,仍需在反应中心添加少量显性水分子;计算出的绝对电位值依赖于DFT计算功函数的精度,目前仍有系统误差。
第一性原理如何研究Tafel动力学
第一性原理计算不再是独立的理论工具,而是成为了解释和预测Tafel动力学的核心手段。它通过揭开反应过程的“黑箱”,为研究人员提供了分配反应机理、理解斜率值、乃至设计高性能催化剂的定量物理基础。
表2. 第一性原理如何服务并深化Tafel动力学分析
基于第一性原理计算出的电荷转移系数(α)的值,将表面电催化反应分为三类。α值直接反映了反应能垒对电位的敏感程度,是连接电子转移与原子重排(化学键断裂/形成)的关键微观物理量。
第一类:电子转移与化学反应强耦合(α≈0.5)
特征:这类反应的α值接近0.5。这意味着反应能垒高度强烈地依赖于外加电位,电位每增加1V,能垒大约下降0.5 eV。这是典型的“电位驱动”的反应。
机制:属于Eley-Rideal机制。反应物之一来自溶液(通常是水合质子H3O+(aq)或OH–(aq)),它直接在表面上与另一个吸附物种发生反应。
电子转移步骤与质子转移或化学键的断裂/形成步骤是协同、同步发生的,是一个完整的“质子–电子耦合转移”(PCET)过程。
第二类:电子转移与化学反应解耦(α≈0)
特征:这类反应的α值接近0。这意味着反应能垒几乎不随外加电位变化。电位不是主要的驱动力,反应速率主要由热力学(温度)控制。
机制:属于Langmuir-Hinshelwood机制。所有反应物都已预先化学吸附在催化剂表面上。反应是两个吸附物种之间的重组或分解。这是一个纯粹的“表面化学反应”,电子转移过程在反应前或反应后已经完成,而不在决速步中发生。
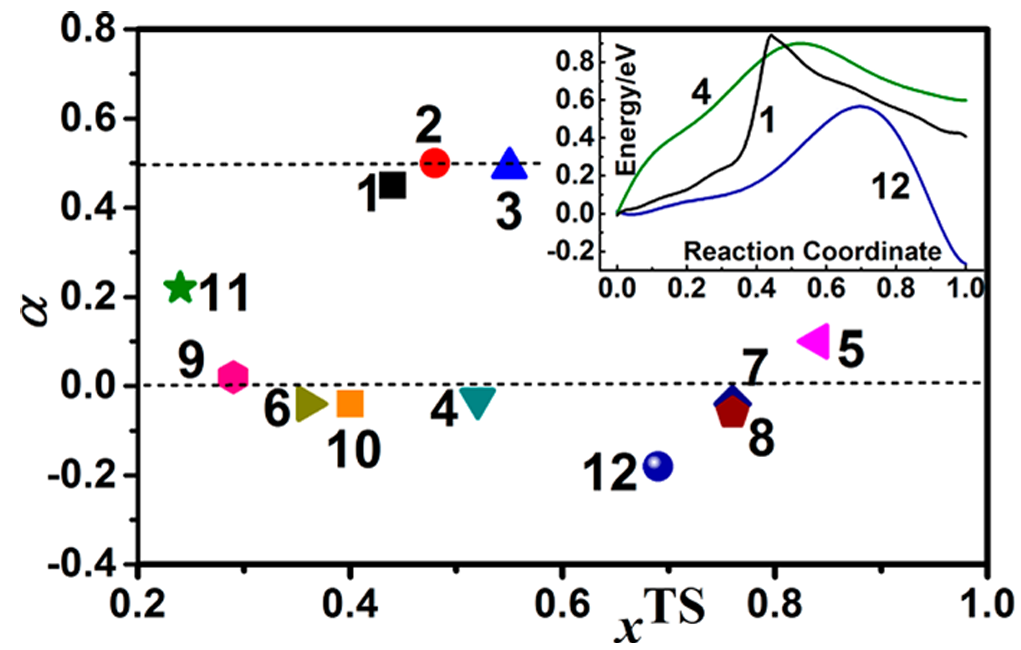
图6. 电荷转移系数(α)与反应坐标(x^TS)的关系散点图。DOI: 10.1021/jp411531f。
第三类:非常规行为(α为非常规值)
特征:这类反应的α值既不是0.5,也不是0,而是一些非常规的值(它们的行为无法用传统的电化学假设来解释。
机制:这类反应揭示了溶剂化壳层结构在反应中的关键作用。反应通常涉及反应物或过渡态与多个水分子形成的特定氢键网络。
从初始态到过渡态,溶剂壳层的结构和极化发生了剧烈且不对称的重组,这种重组与电子转移耦合,产生了一个独特的、非经典的电位依赖性。
总结
本文系统解析“塔菲尔动力学”实验规律与“第一性原理计算”的最新结合方式。Tafel方程虽已有百年历史,仍是电化学动力学分析的核心工具。
未来挑战包括:更精确的电位标定、缺陷表面与纳米颗粒的模拟、多尺度动力学建模、以及光电催化反应的模拟等。随着实验与理论的进一步融合,电催化反应的设计与优化将迎来新的突破。
【做计算 找华算】
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???