




什么是AIMD


AIMD(ab initio molecular dynamics,第一性原理分子动力学)是把电子的量子力学计算直接嵌入到分子动力学的每一步中。传统分子动力学靠预先设定的经验力场给原子“推力”,而AIMD在每个时间步实时求解电子结构(通常用密度泛函理论),由电子分布决定原子受力并推动原子运动。下图提供了AIMD领域发展历程的统计图表(1970-2000年),该图直观反映AIMD方法自Car-Parrinello奠基后的学术影响力增长。

DOI: 10.1021/acsami.2c01347
简单说,传统MD像按菜谱做菜,AIMD则是在烹饪时实时试味并调整配方。正因为直接处理电子,AIMD能自然描述化学键的断裂与形成、电子重排、氧化还原和电荷局域等现象,这些往往是经典力场难以准确表示的。比如在电池电极/电解质界面上,AIMD能揭示电荷如何在原子尺度迁移,从而帮助理解快速充电或界面稳定性问题。
AIMD适用与限制
从计算化学角度,AIMD是研究电子主导过程的利器——如催化反应机理、质子传递、溶剂化壳结构、电极/电解质界面、缺陷诱导的局域态和材料相变等。优点是基于第一性原理,物理基础清晰,结果通常更可信;缺点是计算代价高昂:每个时间步都要做电子结构求解,导致可处理的体系规模通常在几十到几百个原子,时间尺度常在皮秒到几十皮秒。
对初学者的建议是把AIMD当成高精度验证工具或研究小尺度关键过程的“显微镜”,而不是直接取代经典MD。常见的折衷包括先用经典MD或低成本方法做预热与构型筛选,关键段落再用高精度AIMD;或采用QM/MM混合方法把反应区用量子力学处理,其余用经典力场。初学者应从小体系、短轨迹开始,逐步增加复杂度,并详细记录参数设置以便复现与比较。
计算化学研究SEI膜
在计算化学里,AIMD的应用非常广泛且贴近实验可观测量:
研究催化反应路径与过渡态动力学(直接看到键的断裂/形成和中间体);
溶剂化与配位壳的动力学(揭示溶剂分子如何随时间重新排列影响反应);
质子传递和Grotthuss机制(质子跃迁与溶剂重组耦合);
电荷转移与电荷局域化(理解电子如何在分子或材料中迁移或被俘获),下图展示了使用AIMD来研究电解液体系的断键的时间以及溶剂化结构。
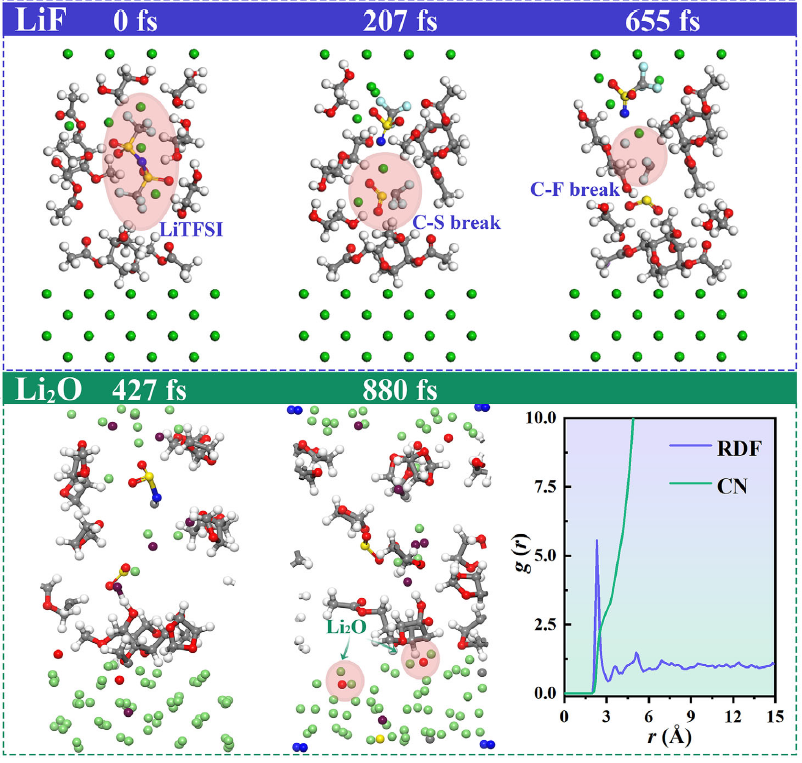
DOI:10.1002/aenm.202502589
电化学界面问题,
如电极/电解质界面的电荷双层、固体电解质界面(SEI)生成与稳定性;
表面吸附与表面反应(腐蚀、表面重构、吸附能随温度变化);
缺陷、掺杂与相变的原子尺度动力学(如何随热扰动演化);
振动光谱模拟(通过轨迹计算红外与拉曼谱线,便于与实验对比);
自由能面与动力学(结合如metadynamics可获得反应自由能与速率);
生物体系的活性位点化学(用QM/MM在酶催化或质子通道中模拟关键事件);
光化学与非绝热过程(使用相应的非绝热AIMD方法可研究电子激发后的动力学)。
下图展示了使用AIMD来研究空气–水界面的非催化水解机理和动力学,通过键长以及自由能等形式来量化。
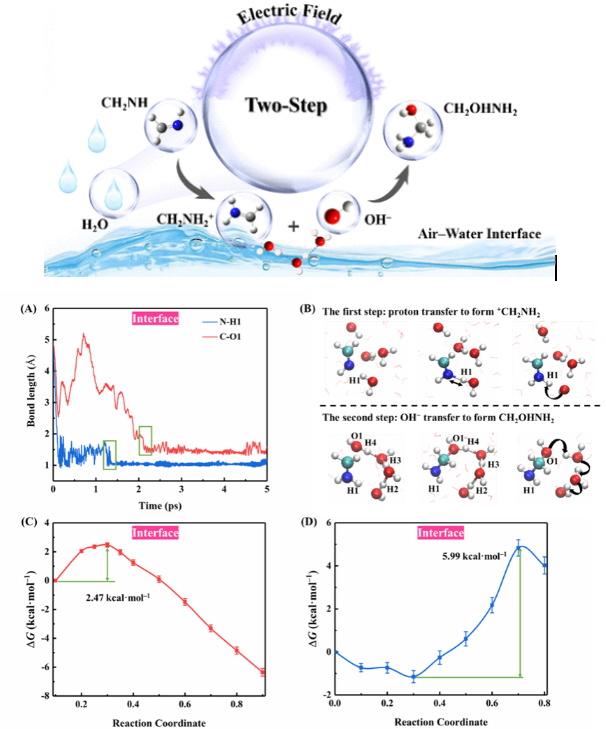
DOI: 10.1021/jacs.4c09080
此外,AIMD常用于验证和改进经验力场:把AIMD的能量、力或统计量作为训练或对照数据,从而提高经典模拟的可信度。实践上,合理选取泛函与赝势、控制时间步和温度耦合、分阶段规划(预热→采样→高精度复核)并结合QM/MM或加速方法,是把AIMD用好、把成本降下来的关键。
总之,AIMD在计算化学中既是理解微观化学机制的强大工具,也是连接电子结构与动力学测量的桥梁;初学者从小体系、具体问题入手,逐步掌握设置与分析,就能把这把“电子显微镜”用得顺手。
总结
AIMD把电子结构直接带入动力学模拟,能真实描述键断裂、电荷转移与界面反应。实践要在精度与成本间取舍:选好DFT泛函与赝势,时间步约0.5–1fs,热浴控制温度;可先用经典MD或低精度DFT筛构型,关键阶段用AIMD或QM/MM等拓展尺度。初学者从小体系、短轨迹开始,学习能量/力收敛、轨迹分析(RDF、配位数)及电子性质提取。分阶段验证使结果更可靠,AIMD是连接电子结构与动力学的重要工具。