

说明:本文华算科技系统介绍了粗粒化分子动力学(CGMD)的基本概念、主要方法及其在生物、材料和药物领域的应用。通过学习本文,读者能够了解CGMD在多尺度模拟中的重要作用,掌握Martini力场、力匹配方法和机器学习辅助粗粒化等核心技术,并认识到这些方法在模拟大分子体系、预测材料性能和药物设计中的价值。



什么是粗粒化分子动力学
粗粒化分子动力学(Coarse-Grained Molecular Dynamics,简称CGMD)是一种介观尺度的分子模拟方法,通过将多个原子或分子分组为单个“珠子”或颗粒,来简化体系的自由度,从而实现对更大空间尺度和更长时间尺度的模拟。
与全原子分子动力学(All-Atom MD)相比,CGMD大大降低了计算复杂度,能够处理数百万颗粒的体系,适用于研究生物膜、聚合物、药物载体和纳米材料等复杂系统。CGMD的核心思想是保留体系的关键物理化学特性,如疏水效应、氢键和静电相互作用,同时忽略次要细节,以提高模拟效率。
DOI: 10.1038/s41524-019-0261-5
CGMD的多尺度建模从底部向上(bottom-up)和顶部向下(top-down)两种策略出发。底部向上基于原子级数据推导粗粒化力场,而顶部向下则通过匹配实验属性优化参数。
不同应用场景下,CGMD的重点有所差异:生物膜模拟关注脂质排列和渗透性,聚合物模拟聚焦链缠结和机械性能,而药物设计强调分子吸附和释放动力学。计算结果受力场选择、颗粒映射和时间步长的影响。随着机器学习的融入,CGMD的准确性和适用性进一步提升。
粗粒化分子动力学的方法
CGMD在分子模拟中应用广泛,涵盖多种粗粒化策略,包括力场开发、颗粒映射和动态整合等。这些方法从不同角度简化体系,助力复杂行为的预测和材料优化。
1、粗粒化模型的构建
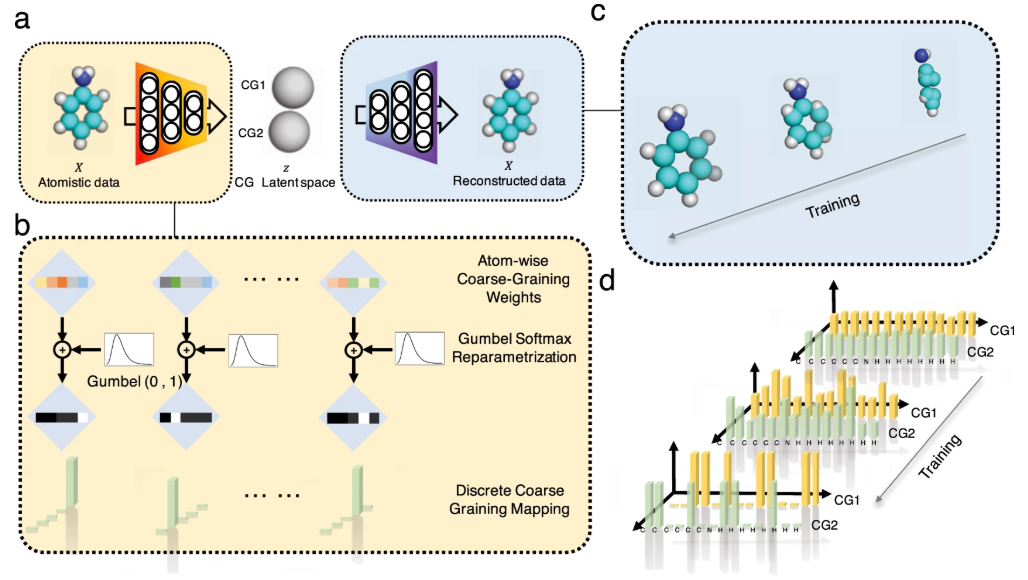
粗粒化模型的构建是CGMD的基础,通过将原子群映射为粗粒化颗粒来减少自由度。典型映射规则如“四合一”(四个重原子对应一个颗粒),分为极性、非极性、近极性和荷电类型,以保留化学特异性。在蛋白质模拟中,粗粒化模型可分为单点模型(每个氨基酸一个颗粒)和多点模型(每个氨基酸多个颗粒),后者更精确但计算量较大。
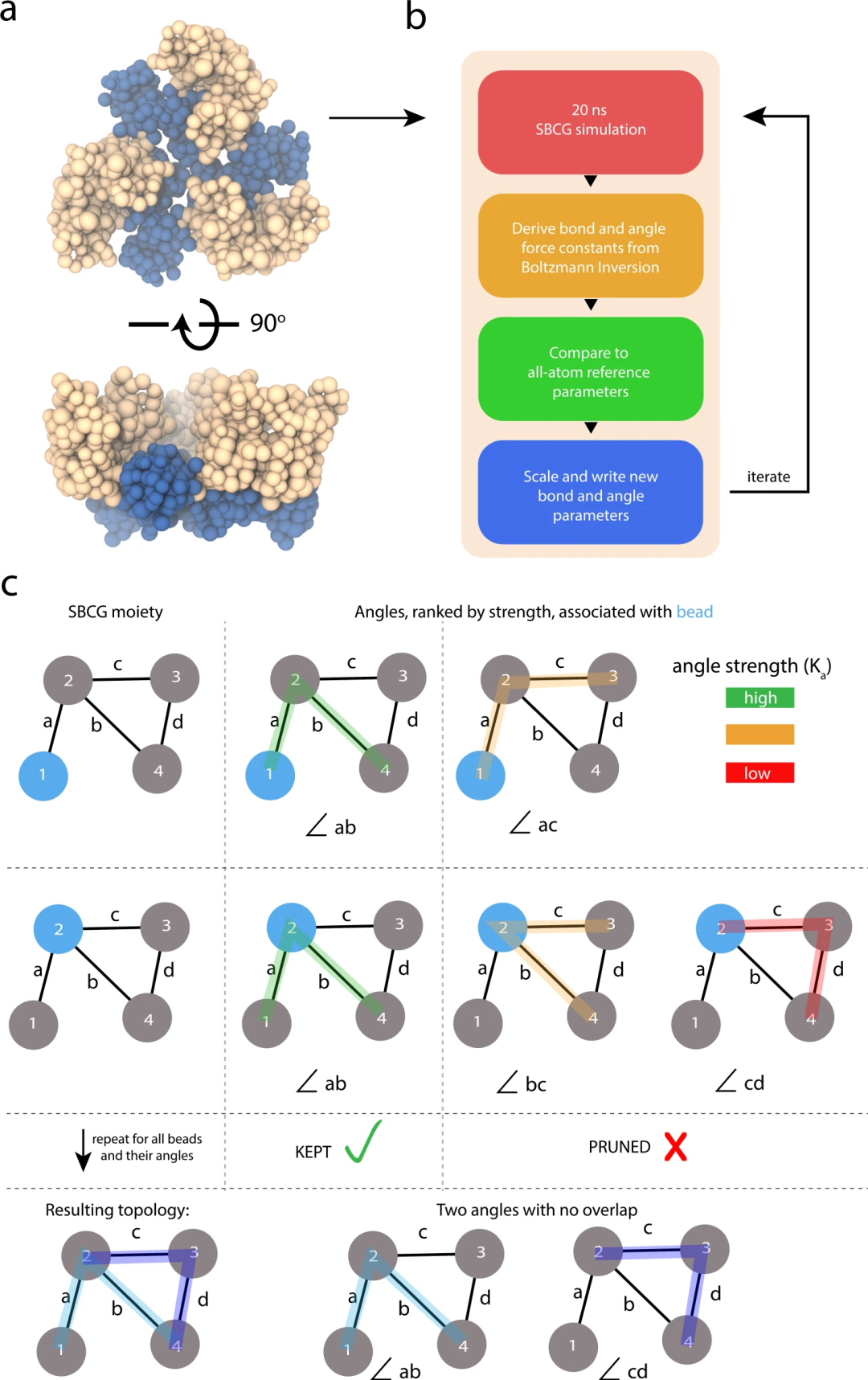
DOI: 10.1038/s41467-023-37801-5
例如,有学者展示了基于形状的粗粒化(SBCG) 方法的重大进展,我们将其称为 SBCG2,其利用代表网络的拓扑的重新制定的公式,使高粒度建模成为可能,并保留维持装配特性的原子细节。此外,提出了一种基于电荷密度傅里叶壳相关性的粒度选择方法,并另外开发了一种细化方法来优化、调整和验证高粒度模型。
2、Martini力场及其应用
Martini力场是CGMD中最常用的力场之一,专为生物分子设计,支持脂质、蛋白质和DNA等体系的模拟。它采用简化的相互作用形式,结合隐式溶剂模型,适用于大尺度体系如脂质双层和胶束。
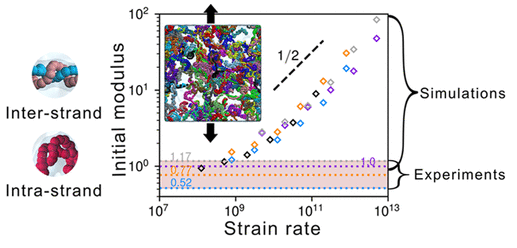
DOI: 10.1021/acsami.4c18162
Martini力场在研究药物载体和膜渗透性方面表现优异,例如MARTINI 水凝胶是使用全原子分子动力学 (AAMD) 通过迭代玻尔兹曼反转 (IBI) 进行分子开发的,并通过目标水凝胶的实验实现来评估其质量。开发的模型提供了一种机械高保真 CG 水凝胶,可以访问大规模的含水水凝胶行为,这在实际时间中很难通过 AAMD 进行探索。
通过建模的水凝胶,我们揭示了聚合物构象调节了水凝胶的弹性从折叠状态到肿胀状态,Panyukov 模型证实了这一点。结果为将聚合物构象和排列与其整体变形联系起来提供了一个强大的桥梁,从而能够实现水凝胶应用所需的多方面和材料特异性预测。
3、机器学习辅助粗粒化
机器学习(ML)在CGMD中用于开发力场和预测动态行为,通过深度学习优化多体相互作用,提升模拟精度。ML结合全原子数据和实验结果,可加速新材料发现和药物设计。
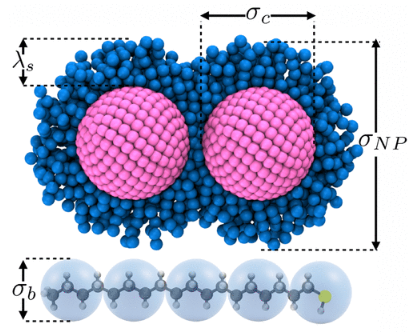
DOI: 10.1021/acsnano.3c04162
例如,ML- CGMD中提出的多尺度方法构成了一种通用的自下而上的粗粒度策略,其中作用在粗粒度位置上的粗粒度力是通过测量参考细粒度模拟中这些位置上的矢量平均力来提取的。这些有效的粗粒度力,即平均力或自由能表面的势的梯度,由结构描述符的梯度的简单线性模型表示,结构描述符是旋转不变的标量函数。
这样,我们也直接得到了粗粒模型的自由能面作为所有粗粒坐标的函数。我们期望这种简单而准确的平均力多体势的粗粒度框架将能够通过直接模拟来表征、理解和预测相关软物质系统的结构和相行为。
结论
粗粒化分子动力学(CGMD)通过模型简化、Martini力场和机器学习等方法,从介观尺度研究复杂体系的动态行为,为生物、材料和药物领域提供高效工具。GCMD在模拟生物膜、聚合物和纳米载体方面展现出巨大潜力,推动了精准医学和材料设计的进步。随着计算技术和算法的发展,CGMD将在多尺度模拟和跨学科研究中发挥更大作用。
【做计算 找华算】