

说明:本文华算科技系统阐述分子模拟中力场技术的核心概念、分类体系及选择原则。力场作为分子模拟的数学基础,通过经验参数化方法描述原子间相互作用,成为连接量子力学与宏观现象的关键桥梁。本文将深入分析力场的基本原理、发展脉络和实际应用策略,为研究者提供全面的技术参考框架。



什么是力场?
力场(Force Field)是分子模拟领域的核心计算框架,本质上是一套数学表达式与参数集合,用于描述分子系统中粒子坐标与势能之间的函数关系。在分子动力学(MD)、蒙特卡罗(MC)以及量子力学/分子力学(QM/MM)混合模拟中,力场通过一组经验或半经验的势能函数,将分子结构、能量和动力学信息转化为可计算的形式。
从物理本质来看,力场采用势能函数分解的策略,将体系的总势能Etotal表达为多个相互作用项的加权求和:
Etotal=Ebonded+Enon-bonded=(Ebond+Eangle+Etorsion)+(EvdW+ECoulomb)
其中键合项(bonded)描述分子内共价相互作用,包括键长拉伸、键角弯曲和二面角扭转能;非键合项(non-bonded)则涵盖范德华力、静电相互作用等分子间作用力。
力场参数的获取是一个多源数据融合过程:通过拟合实验观测数据(如晶体结构、光谱学数据)、高精度量子力学计算或半经验方法获得。这种参数化策略使力场既保持了物理准确性,又实现了计算效率的平衡,成为连接微观量子世界与宏观实验现象的关键桥梁。
DOI: 10.1021/acs.jctc.9b01190
力场的分类
力场体系经过数十年发展,已形成多维度分类标准,可根据物理模型、应用领域和参数化策略进行系统划分。
1、按物理模型复杂度分类
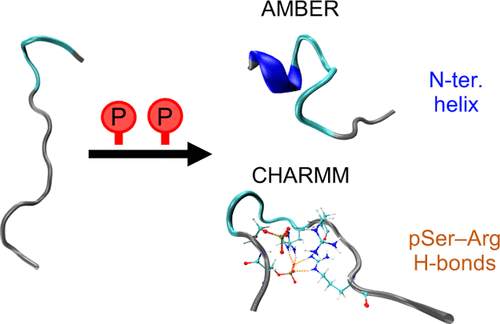
传统固定电荷力场是最成熟的应用体系,包括AMBER(主要针对生物大分子)、CHARMM(兼顾生物与材料体系)、OPLS(侧重有机液体)、GROMOS(专注于凝聚相系统)以及DREIDING(通用材料力场)等。这类力场采用原子中心点电荷模型,计算效率高但无法描述电子极化效应。
极化力场通过引入可极化偶极子或浮动电荷,显著提高了对电子分布变化的响应能力。包括Drude振荡器模型、诱导偶极矩模型和电荷平衡方法等。这类力场能更准确地模拟界面体系、离子溶液和激发态过程,但计算成本增加3-5倍。
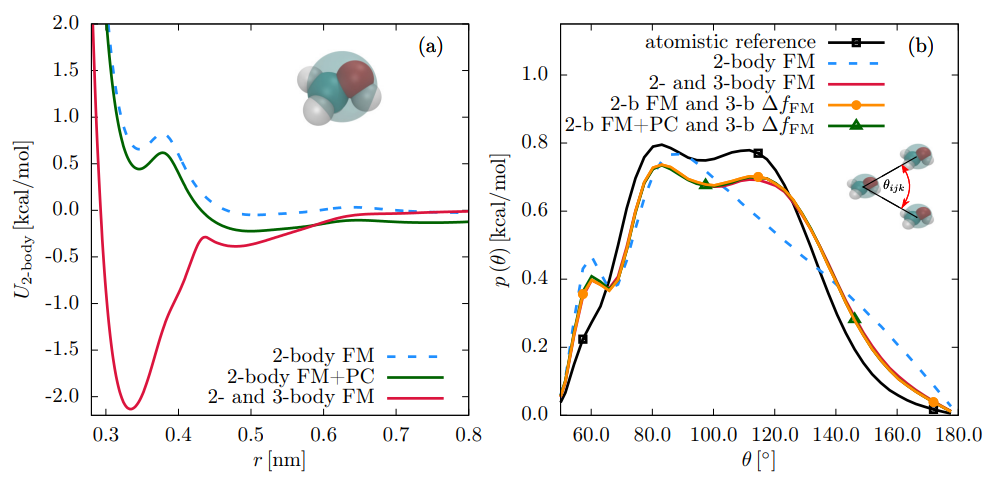
粗粒化力场采用珠簧模型将多个原子映射为一个相互作用位点,极大提升了时空尺度模拟能力。MARTINI力场是典型代表,可将模拟尺度推进到微秒级和微米级,适用于生物膜自组装、高分子相分离等介观现象研究。
2、按参数化策略分类
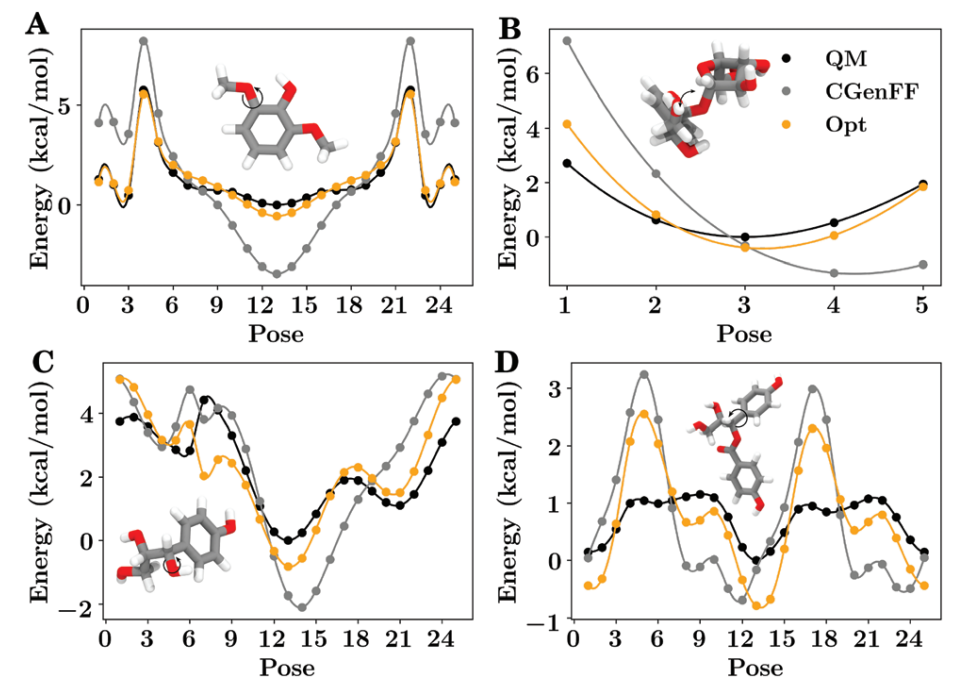
经验力场主要依靠实验数据进行参数拟合,如振动频率、构象能差和液态性质等。GROMOS和早期OPLS力场采用此策略,物理基础扎实但参数转移性较差。
DOI: 10.1039/c8gc03209b
量子力学衍生力场基于第一性原理计算数据构建参数集,包括GAFF(通用AMBER力场)、MMFF94等。这类力场具有更好的通用性和可转移性,尤其适用于新型材料分子的模拟。
机器学习力场是近年兴起的革命性技术,通过神经网络学习量子力学计算数据,能保持量子精度同时提升计算效率2-3个数量级。如ANI、SchNet、DPMD等模型已在催化反应、材料设计等领域展示强大潜力。
3、按应用领域分类
生物分子力场专门针对蛋白质、核酸、脂质等生物体系优化,包括CHARMM36、AMBER19SB、GRORMOS54A7等。这些力场在二级结构稳定性、膜蛋白折叠和蛋白质–配体结合能计算方面经过特殊参数化。
材料力场面向无机材料、金属有机框架(MOF)和纳米材料设计,如COMPASS、UFF、Dreiding等。特别在表面吸附、机械性能和热传输等方面进行重点优化。
反应力场突破传统力场不能描述化学键断裂形成的限制,如ReaxFF通过键级势函数实现反应过程的动态模拟,在燃烧过程、材料断裂等研究中不可替代。
如何选择力场
力场选择需遵循系统特异性、精度要求与计算成本三重平衡原则,具体决策流程需考虑以下维度:
1、研究体系匹配原则
对于生物大分子体系,首选AMBER、CHARMM或GROMOS系列力场。若研究涉及蛋白质–配体结合,推荐使用AMBER19SB+GAFF组合;膜蛋白模拟宜采用CHARMM36力场与SLIPID膜模型搭配;糖类分子则推荐GLYCAM力场。
DOI: 10.1039/c8cp00746b
材料科学领域需根据材料类型细分:金属体系适用EAM或MEAM势函数;碳材料选择AIREBO或ReaxFF;MOF材料推荐UFF或Dreiding;聚合物材料则适用PCFF或COMPASS力场。
溶液体系的选择取决于溶剂性质:水溶液可采用TIP3P、TIP4P等水模型;离子液体需使用专门参数化的力场如CL&P;有机溶剂宜选择OPLS系列力场。
2、精度与效率权衡策略
当研究平衡性质(如密度、扩散系数)时,传统固定电荷力场通常足够;
若涉及介电响应、界面效应或激发态过程,则必须采用极化力场;
对于化学反应或键断裂过程,反应力场是唯一选择。
时间尺度也是关键考量:纳秒级模拟可采用全原子力场;微秒级模拟建议使用粗粒化力场;毫秒级以上模拟则需采用超粗粒化或连续介质模型。
空间尺度同理:小体系()可用全原子模型;大体系(>100 nm)需采用多尺度方法。
3、参数化质量评估标准
优秀的力场应通过可转移性验证、温度压力稳定性测试和实验数据对标三重检验。需特别检查:
①参数是否源于高质量量子计算或实验数据;
②是否经过热力学积分验证;
③是否在类似体系中有成功应用案例。
建议采用渐进式验证策略:先计算分子构象能曲面与量子结果对比;再模拟简单体系的热力学性质与实验对照;最后进行目标性质的扩展模拟。同时注意力场参数的版本兼容性,不同版本的参数混合使用可能造成系统性误差。
4、新兴技术融合应用
机器学习力场虽具潜力但目前仍需谨慎使用。建议采取混合策略:用机器学习力场进行采样,再用传统力场进行分析;或采用主动学习方案,在模拟过程中动态优化力场参数。
小结
力场技术作为分子模拟的数学基石,通过精巧的参数化策略平衡计算效率与物理精度,使原子尺度模拟成为探索物质科学规律的核心手段。从传统固定电荷力场到可极化模型,再到机器学习力场的演进,体现了计算科学从经验参数化向数据驱动范式的转型。
未来力场发展将呈现多尺度融合、智能化参数化和专用化定制三大趋势,为精准分子设计提供越来越强大的计算引擎。选择适用力场需坚持系统适配性原则,结合研究目标和计算资源做出最优决策,同时保持对力场局限性的清醒认知。
【做计算 找华算】
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服
务。?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???