




什么是经典分子动力学和从头算分子动力学
在分子动力学(MD)世界里,所有原子的运动都被牛顿第二定律所支配,核心任务是计算出每个原子受到的力。传统的分子动力学,也叫经典分子动力学,使用预先设定好的“力场”来描述原子间的相互作用。
这些力场就像是经验公式,告诉计算机在某种距离下两个原子应当互相吸引或排斥多大。常见的力场有AMBER、CHARMM、OPLS等,它们通过实验数据或高层量子化学计算拟合得到,因此在常规分子、蛋白质或材料模拟中既准确又高效。
相比之下,从头算分子动力学(AIMD, Ab Initio Molecular Dynamics)不依赖经验力场,而是实时求解量子力学方程(通常是密度泛函理论 DFT)来计算电子结构,从而得到原子间的力。
简单地说,传统MD只看原子“壳”的运动,而AIMD还考虑了电子“心”的响应,这让它在描述化学反应、键断裂与成键、极化、电荷转移等量子效应时更为准确。
下图直观地对比了AIMD与经典MD在可模拟的体系尺寸和时间尺度上的巨大差异。该图说明了AIMD通常只能模拟数百个原子在皮秒尺度内的动力学(图a),而经典MD利用经验力场可以模拟数百万个原子在微秒尺度的行为(图b)。
DOI: 10.1021/acs.jpcc.8b09917
两者区别
从计算化学的角度看,AIMD和MD的主要区别在于“精度换效率”。
AIMD的最大优点是物理真实感:它不需要预设力场,能自洽地描述电子与原子的相互作用,因此适合研究反应机理、电极电解质界面、电荷转移、金属催化等体系。
然而,这种高精度的代价极为昂贵——一次AIMD模拟的时间步通常是飞秒级别,而模拟的总时长往往只到皮秒量级,体系原子数也通常限制在几百个以内。
相比之下,经典MD由于使用解析形式的力场方程,计算效率极高,可以轻松模拟上百万个原子、纳秒甚至微秒级时间尺度的过程,如蛋白折叠、离子扩散、纳米通道流动等。
因此,在科研实践中,研究者往往根据研究对象来权衡选择:
如果研究的是结构动力学或统计热力学性质,用MD;
若研究的是化学反应、电子重排或极化效应,则必须使用AIMD。
也有中间路线——机器学习势函数(ML Potential)结合了AIMD的精度和MD的效率,成为当下热门的发展方向。
下图系统地展示了机器学习势函数(MLP)的工作原理。其核心思想是,MLP通过从AIMD计算中学习,力图在保持接近AIMD精度的同时,获得接近经典MD的计算效率,从而在精度与效率之间架起桥梁。
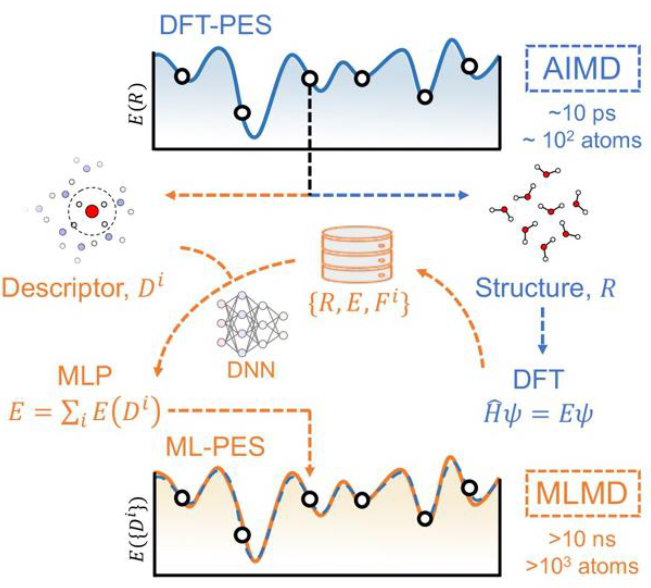
DOI:10.1039/B802376J
互补关系
在现代计算化学工作流中,AIMD和MD常常并非对立,而是互补。
研究者可以先用AIMD对关键反应位点或电子效应进行高精度建模,再将得到的势能面或力场参数转移到经典MD中进行大尺度模拟,从而同时兼顾微观精度和宏观统计效果。
这种多尺度方法在电解质、界面、催化反应、固液边界等研究中尤为重要。
例如,在研究电极/电解液界面时,AIMD可以准确计算溶剂化壳、离子配位和电子转移,而MD则能模拟更长时间的离子输运与界面稳定性。
下图通过AIMD模拟快照和电子性质(如态密度和能带结构),展示了碱金属-氨溶液中溶剂化电子的分布和电子转移行为。
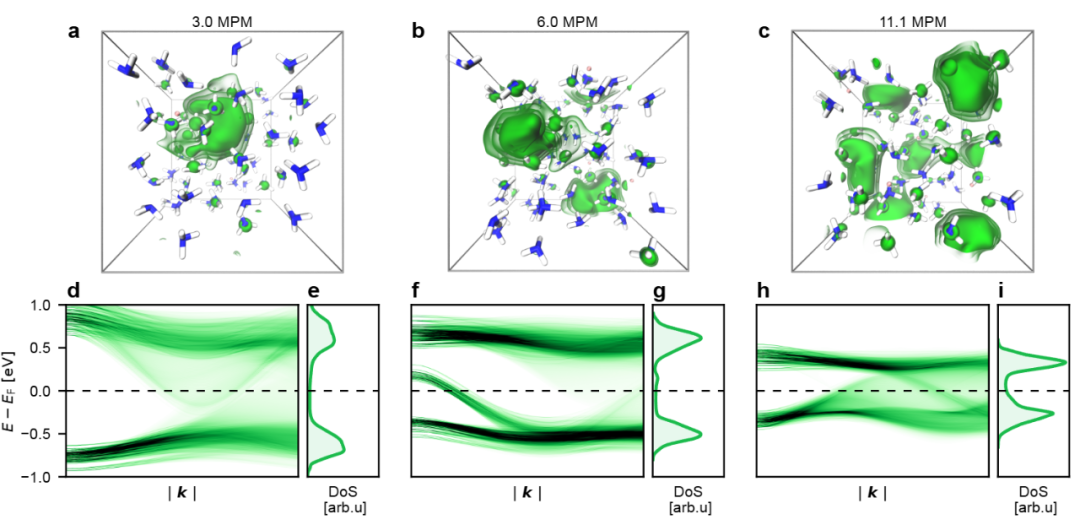
DOI:10.26434/chemrxiv-2024-xpzd0
下图通过AIMD模拟展示了聚电解质–离子系统中第一水合壳的分子可视化结构,清晰呈现了离子配位和溶剂化壳的细节,比了AIMD和经典MD模拟的径向分布函数(RDF),突出了AIMD在描述溶剂化壳结构方面的准确性。
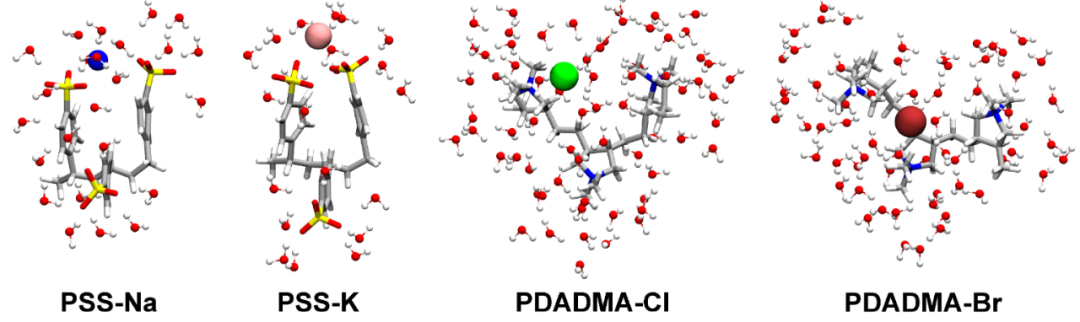
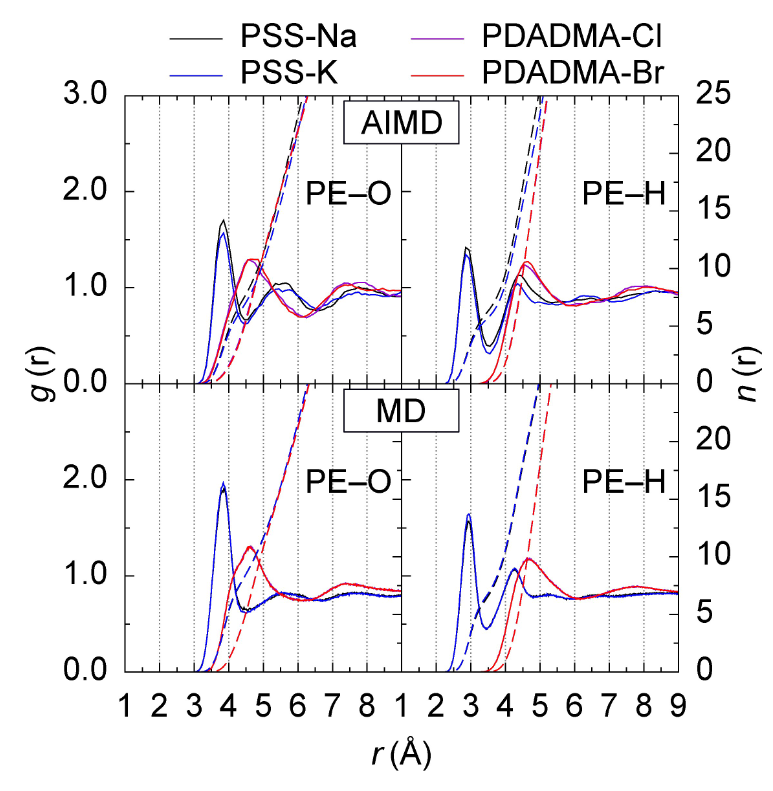
DOI:10.1002/cphc.202400244
近年来,随着高性能计算和人工智能算法的发展,AIMD的计算速度不断提升,经典MD的力场也在不断量子化、极化化,二者的边界正在变得模糊。未来的趋势是通过机器学习自动从AIMD数据中生成势能模型,实现“量子精度、经典速度”的统一,这将彻底改变计算化学的研究范式。
总结
AIMD与MD是分子模拟中两种不同层级的计算方法,一个追求量子精度,一个强调时间与空间尺度。
AIMD能揭示反应机理、电子结构变化与极化效应,是研究微观化学本质的重要工具;MD则以高效计算著称,能捕捉复杂体系的动力学演化与热力学统计特征。
实际研究中,二者常结合使用——AIMD提供精确的局域信息,MD负责延展到更大体系与更长时间。随着计算资源和机器学习势函数的发展,AIMD与MD的界限正逐渐模糊,多尺度、数据驱动的模拟框架正在成为计算化学的新趋势。理解二者的原理与适用范围,是迈入现代分子模拟的关键第一步。
【做计算 找华算】
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???