

HOF依靠氢键作用自组装成多孔结构,相较MOF和COF更轻盈灵活,具备良好的可逆性和环境友好性。从计算化学角度,研究者利用DFT与分子动力学等方法,探索其稳定性、孔隙率和吸附性能,为气体分离、碳捕获及药物递送等领域提供理论支撑。
在材料科学的世界里,有一种新兴的“轻量级选手”正在受到越来越多的关注,它们叫做HOF材料,全称是Hydrogen-bonded Organic Frameworks,中文常译为氢键有机框架材料。顾名思义,这类材料是通过分子之间的氢键作用自组装而成的三维多孔结构。下图展示了HOF-29材料的AA堆叠(垂直与侧面视图)和AB堆叠模式(垂直与侧面视图)。
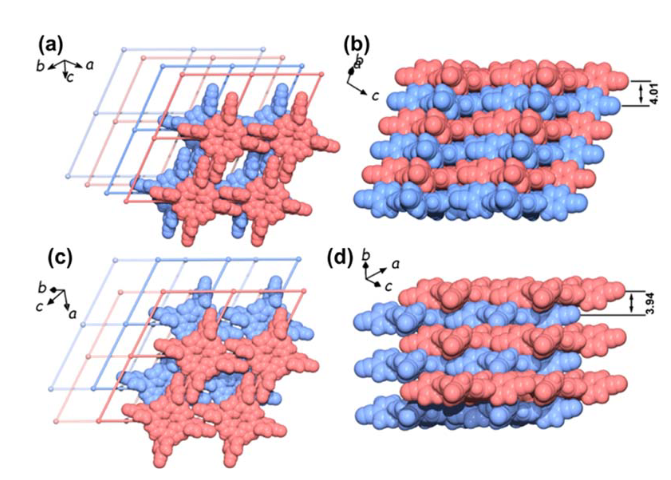

DOI:10.1039/D4SC02628D
相比于我们更常听到的MOF(金属有机框架)和COF(共价有机框架),HOF最大的特点是:它的“搭建方式”更温和,主要依靠氢键这种弱相互作用,而不是金属–配体配位键或强共价键。
也正因为氢键的柔性,HOF结构往往更加轻盈,可逆性更好,甚至在某些条件下可以重新组装。对初学者来说,可以把HOF理解成内部有许多孔洞,可以用来装气体分子、离子甚至药物分子的多孔结构。
那么,从计算化学的角度,我们如何理解HOF呢?首先,氢键是一种中等强度的相互作用,比范德华力强,却又比共价键弱,这种“适中”的特性让HOF既稳定又灵活。
在计算中,我们通常会利用量子化学方法(比如密度泛函理论DFT)来分析单个氢键的能量、方向性和电子结构,从而预测整个框架能否稳定存在。下图展示了使用DFT理论来研究HOF与电解液不同组分的吸附能、HOF分子静电势以及电解液在HOF Li 和bare Li上的接触角。
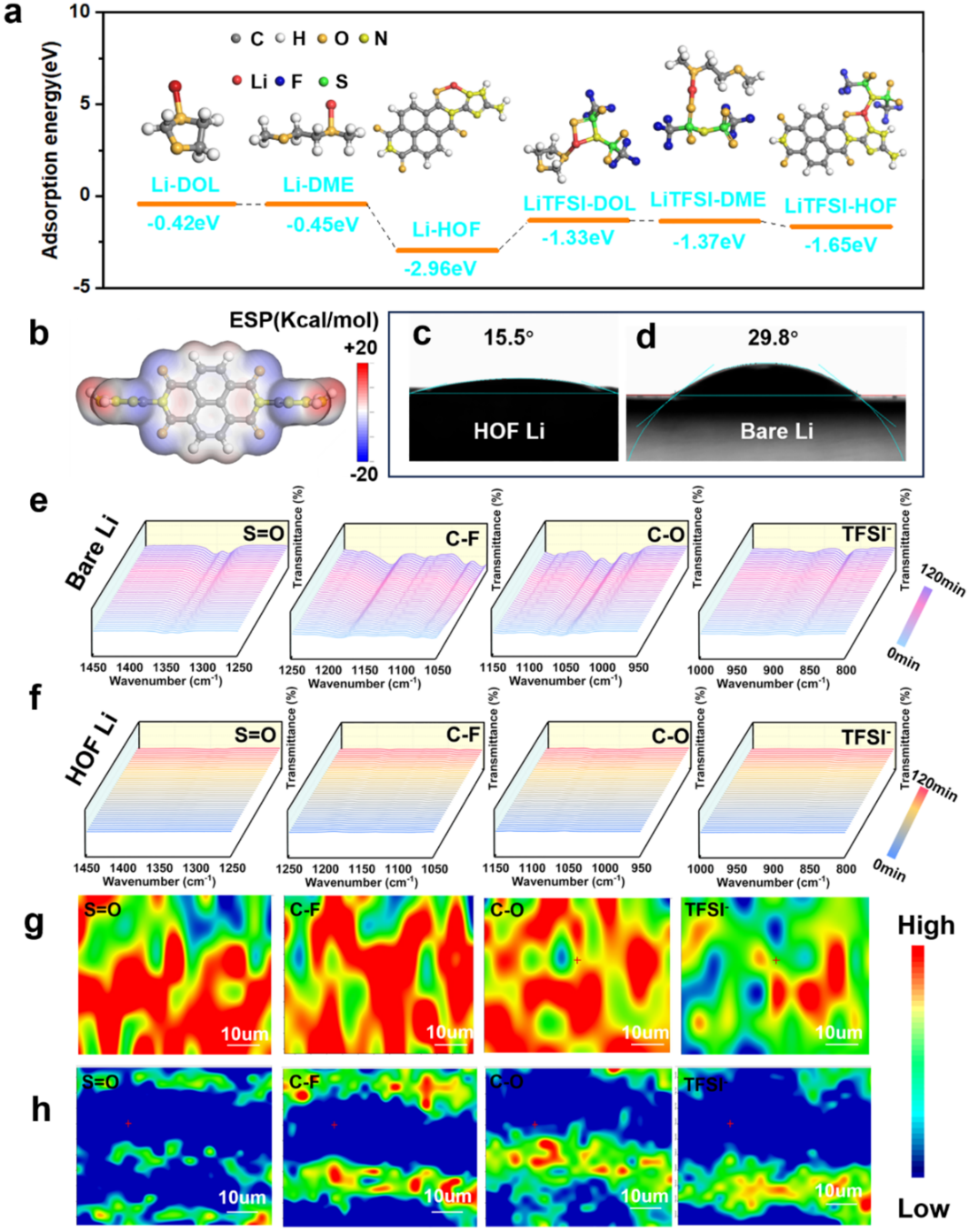
DOI: 10.1002/anie.202506892
下图展示了HOF首次用于锂金属人工SEI的构筑,能够加速锂离子的去溶剂化过程,并引导锂快速且均匀地沉积。
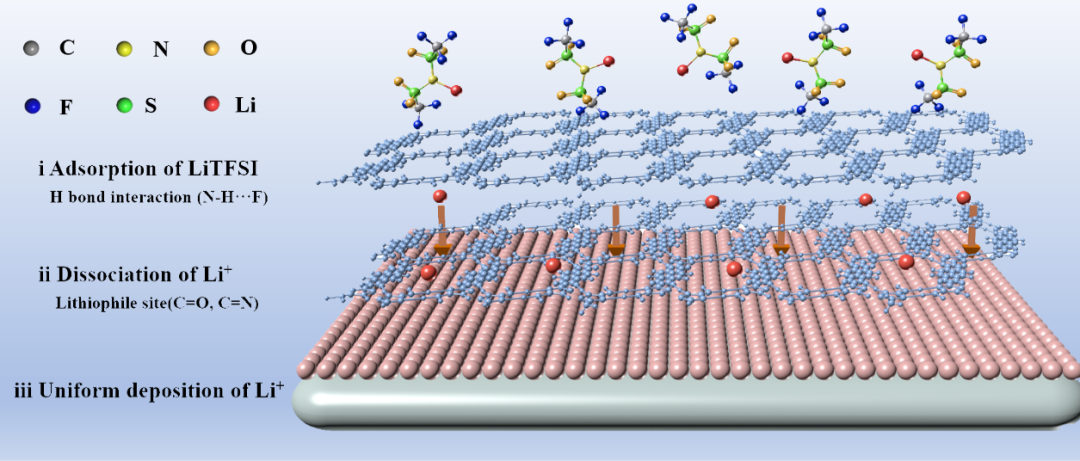
DOI: 10.1002/anie.202506892
另一方面,HOF的孔隙率和分子排列方式可以通过分子动力学模拟来研究,比如模拟气体分子在孔道中的扩散轨迹,或预测在不同温度、压力下框架是否会塌陷。这些计算结果对于实验学者来说,能帮助他们更高效地设计和筛选出性能优良的HOF材料。
比如,有些HOF在模拟中表现出对二氧化碳的强吸附能力,这就意味着它们有可能应用于碳捕获与储存。下图详细展示了CO2分子在HOF-5a孔隙中的有序嵌入排列模式,直观呈现了气体分子在动态变形孔道中的扩散轨迹与空间分布。
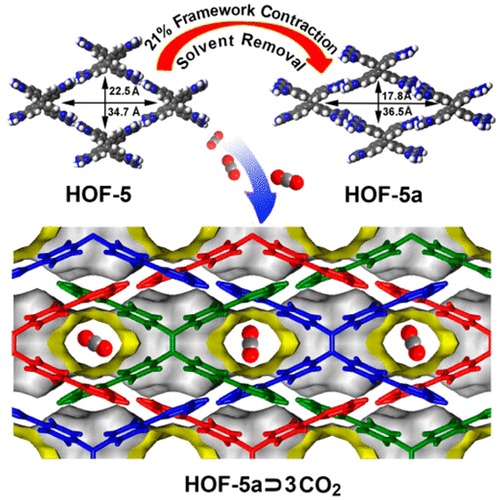
DOI:10.1021/jacs.5b05644
很多人可能会问,HOF既然是靠氢键维系的,那不是很脆弱吗?事实上,正是这种脆弱和可逆性,给HOF带来了独特的优势。首先,HOF在溶液中可以比较容易地组装和重构,这使得它们的合成过程更环保、更节能。
其次,因为分子设计的自由度很大,我们可以通过改变化学基团来“定制”氢键的强弱和排列方向,从而控制孔径大小和化学环境。对于计算化学研究者来说,这就像玩“分子版的乐高积木”:我们可以在计算机上先尝试各种组合,找到既稳定又有应用潜力的结构,再指导实验合成。
未来,HOF有望在气体分离、药物递送、传感器、能源存储等领域发挥作用。相比MOF和COF,HOF更轻量、更灵活,甚至可能在生物医学应用中展现优势,因为氢键的本质更接近生物分子之间的相互作用。
HOF材料作为一种新兴的多孔有机框架,凭借氢键这种“弱而有序”的连接方式,展现出与传统MOF、COF截然不同的优势。它们的结构不仅轻盈可调,还能在外部条件变化时保持良好的重组能力。通过计算化学方法,我们能够从分子尺度深入理解氢键的能量和方向性,预测框架的稳定性与功能表现,为实验设计提供清晰的理论蓝图。
更重要的是,HOF的合成过程通常更加环保,且在药物载体、生物材料和能源应用中拥有广阔前景。随着计算与实验的结合,这类“氢键积木”有望在未来材料科学和可持续发展领域中扮演关键角色。