

说明:本文华算科技主要介绍材料科学中晶体结构的基本含义,以及晶胞、点阵、基元、对称性、晶面和晶向怎样进入第一性原理计算中的结构模型。





晶体结构描述的是原子在三维空间中按周期规则重复的方式。一个晶体模型至少包含两类信息:晶胞矢量给出重复框架,原子坐标给出框架内部的基元。把晶胞沿 a、b、c 方向平移,就能铺成宏观晶体;把基元换掉或移动到不同分数坐标,同一个点阵也会变成另一种材料。
DFT 读取的结构文件并不认识“块状固体”这个笼统对象,它读取的是晶胞矩阵、元素种类、原子数和分数坐标。Sr3AsI3 这类立方卤化物钙钛矿中,Sr、As、I 的空间位置决定了近邻配位和键长;同一张图右侧的布里渊区则来自倒易晶格。实空间晶胞和倒空间路径是一对对象,结构模型改变后,能带路径、声子路径和 k 点采样都会跟着改变。
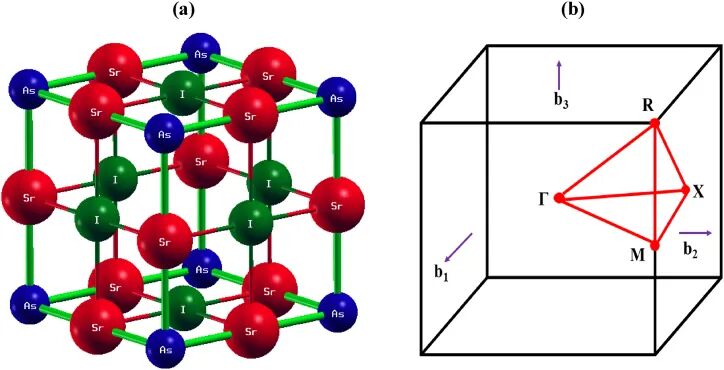
晶体结构读法要避开“球棍图长得像什么”的表面印象。球的颜色和大小只是可视化约定,真正参与计算的是坐标、周期重复关系和原子相互作用。分数坐标靠晶胞矢量定义实际距离,所以晶胞参数变了,同一个 0.25、0.50、0.75 坐标对应的键长也会变。讨论结构时要同时说清材料成分、晶胞参数、原子占位和空间群,孤立的结构截图很难支撑后面的能量或性能判断。
实验晶体学表中常见 0.5、0.75 这类占位数,表示某个 Wyckoff 位置在平均结构中并非总有同一种原子,占位、空位和部分占据会让结构模型更具体。DFT 通常要把平均占位转成确定的原子排布,可能需要有序近似、特殊准随机结构或多个构型比较。从实验结构到计算结构,最容易改变的正是局域配位和短程有序。
点阵是抽象的周期平移集合,基元是附着在每个点阵点上的原子组合。金属、离子晶体、分子晶体和钙钛矿都可以用这套语言描述,但基元大小差别很大。分子晶体的基元往往包含完整分子,晶胞内还可能有多个取向不同的分子;无机晶体的基元常写成若干阳离子和阴离子的配位多面体。

图2中的 L-asparagine monohydrate 说明,晶胞内部可以包含分子取向、氢键网络和水分子位置。对这类体系,DFT 若只优化孤立分子,只能得到分子内键长和电荷分布;周期晶胞计算才会看到氢键、堆积方式和晶格能。基元不是化学式的机械复写,而是晶胞内真实的原子排布。
层状卤化物钙钛矿给出另一类例子。相同的无机层骨架会被有机阳离子隔开,阳离子位置、取向和氢键会改变层间距、极性和磁耦合。同一种组成可以拥有不同晶体结构,结构差异不一定来自元素种类,而可能来自基元取向、层间堆叠和局部对称破缺。

在结构建模中选择原胞、常规胞和超胞时,三类晶胞承担的任务并不相同:原胞原子数最少,适合体相能量、能带和声子计算;常规胞更便于对应晶系、空间群和实验结构表;超胞用于缺陷、掺杂、磁序、声子有限位移或表面模型。超胞不是新材料本身,它是为了容纳局域扰动或降低周期镜像相互作用而扩展的计算盒子。
原胞太小会强迫缺陷周期性重复,掺杂浓度会被人为抬高;超胞太大又会增加 k 点、内存和结构弛豫成本。磁性材料还要考虑铁磁、反铁磁或非共线磁序能否放进当前晶胞。晶胞大小首先限定可表达的物理图案,随后才谈计算收敛和成本。
对称性决定哪些原子等价、哪些张量分量允许存在、哪些能带简并能够保留。空间群把平移、旋转、镜面、反演、螺旋轴和滑移面组织在一起,给晶体结构一个可检索的数学标签。相同空间群仍需检查元素、键长、键角和电子占据;空间群不同也不一定元素不同,很多相变只改变原子位移和局部配位。反演中心是否存在,会直接影响极化、压电和某些自旋分裂判断。

IrTe2 的高温相和调制相给出一个清楚例子:Ir 和 Te 的元素比例没有改变,局部 Ir-Ir 接触和层内重排却能让结构周期扩大,晶胞线框随之改变。对计算来说,若仍用高温小晶胞描述低温调制相,电子稳定、振动熵和相变温度的比较会偏离目标相。多晶型比较必须绑定具体晶胞和原子位置,不能只写同一个化学式。
钒氧化物中,多面体连接方式更能说明对称性和局域配位的作用。VO5方锥、VO6八面体、边共享、角共享、层状骨架和隧道状骨架会形成不同 V2O5 多型。不同连接方式改变 V-O 键强、离子扩散路径和晶体各向异性,DFT 中的能量排序、带隙和迁移势垒都要回到这一套原子连接关系。

结构中保留哪些对称操作,会同时改变计算工作量和弛豫自由度,对称操作越多,不可约 k 点和独立原子位移越少;结构中有缺陷、磁序、吸附物或外场时,对称性会降低,计算量和可比对象都会改变。保留错误对称性会锁住本该弛豫的原子位移,去掉所有对称性又可能让等价位点产生数值噪声。结构优化前后检查空间群和等价原子,是判断模型是否跑偏的必要步骤。
相变、铁电畸变和 Jahn-Teller 畸变都可能从很小的原子位移开始。若优化时强行保持高对称结构,软模对应的位移无法展开,声子谱里可能保留虚频;若允许低对称弛豫,体系可能进入另一个局部极小结构。对称性设置会改变势能面上可访问的位置,这也是结构优化和声子计算需要一起看的原因。
晶向用方括号 [uvw] 表示,晶面用圆括号 (hkl) 表示。晶向描述沿哪个方向看或移动,晶面描述哪一组平行平面切过晶格。单晶力学、离子扩散、电子输运和表面反应都会受方向影响。晶体结构不是各向同性的背景,方向和面指数常常决定实验读到的是哪一套键和迁移路径。

图6中的 (010)、(011)、(100) 等标注把宏观晶体外形和晶格方向连接起来。切 slab 时,同一材料的 (001)、(010)、(101) 表面会暴露不同配位原子,表面能、吸附能和功函数都可能改变;做扩散时,[010] 迁移路径和 [001] 迁移路径的瓶颈半径也可能不同。晶面和晶向是模型条件,不是图注里的装饰。
把同一晶体结构转向体相、缺陷、表面或界面问题时,模型需要保留的结构对象会随目标改变:体相相稳定要用完整周期结构;缺陷需要足够大的超胞和电荷补偿;表面需要 slab、真空层和终止面;界面需要晶格匹配和相对位移;声子和弹性需要确认应力释放。每一种模型都只回答它所保留的结构对象,原胞能带不能替代表面态,理想晶体能量也不能替代缺陷形成能。

图7把光、应力、磁场和衍射信号与晶体方向联系起来。材料科学里说“结构决定性质”,具体到计算语境,指的是晶胞、基元、对称性、晶面、晶向和局域缺陷共同限定哈密顿量。没有结构对象,能量、带隙、声子、吸附和扩散都只是失去参照的数值。晶体结构基础把材料问题压回可计算、可比较、可复核的原子排布。