

说明:本文华算科技介绍了费米能级和d带中心的概念、区别及关联。费米能级是全局性电子化学势,决定电子转移方向;d带中心是局域性轨道属性,反映过渡金属d电子反应活性,影响吸附强度。二者相对位置是决定过渡金属催化活性的关键因素。


费米能级(EF)是描述固体(特别是金属和半导体)中电子集体行为的一个中心概念。它的定义源于费米–狄拉克统计(Fermi-Dirac statistics),该统计描述了费米子在热平衡状态下是如何占据量子态的。

图1 失配应变与温度对费米能的影响。DOI:10.48550/arXiv.2004.05338
在绝对零度(0 K)时,费米能级是电子占据的最高能量量子态的能量。低于的所有能级都被电子填满,而高于EF的所有能级都是空的,清晰地划分了已占据态和未占据态。
在任何非零温度T > 0 K时,费米能级被定义为电子占据概率为50%的那个能量位置。其数学表达式由费米–狄拉克分布函数给出:

其中,E是能量,kB是玻尔兹曼常数,T是绝对温度。当E=EF时,指数项为e0=1,因此f(EF)=1/2。
在有限温度下,费米能级附近的电子会被热激发到更高的能级,导致EF周围出现一个能量宽度约为的“模糊区域”,其中电子占据概率从接近1平滑过渡到接近0。

图2 不同温度的费米–狄拉克分布
从更根本的热力学角度看,费米能级是体系中电子的化学势(μ),即在保持温度和体积不变的情况下,向体系中增加一个电子所需的能量。EF=μ。这个定义更加普适,因为它不仅适用于固体,也适用于任何费米子系统。
作为化学势,费米能级决定了当两个或多个物体接触时电子的流动方向,电子会自发地从费米能级高的物体流向费米能级低的物体,直到它们的费米能级对齐达到平衡。

图3 (a)Co/CoSe2的计算态密度(b)Co/CoSe2异质结接触前后的简化能带示意图。DOI:10.1002/aenm.202502425
费米能级相关定义
功函数(Φ):功函数定义为将一个电子从材料内部的费米能级移到材料外部的真空能级(Evac)所需的最小能量,即Φ=Evac-EF。
功函数衡量了材料束缚电子的能力,它直接影响材料的表面电化学性质和电荷转移能力。在电催化中,催化剂的功函数会影响双电层结构和反应物/中间体的吸附。

图4 功函数(WF)、带隙(EG)、电离能(IE)以及电子亲和能(EA)等相关参数。DOI:10.1039/C5MH00160A
电荷转移基准:在催化剂与吸附物种形成化学键的过程中,电荷会在它们之间重新分布。费米能级作为电子的化学势,是判断电荷转移方向和程度的能量基准。例如,如果一个吸附物的未占据分子轨道(LUMO)能量低于金属的费米能级,电子就倾向于从金属流向吸附物。

图5 Co基催化剂的费米能级图
电催化中的“零点”:在电化学测量和理论计算中,费米能级通常被设定为能量标尺的零点(0 eV)。所有其他电子态的能量,包括核心能级、价带结构以及d带中心,都是相对于其费米能级。

d带中心(εd)是专门用于描述过渡金属电子结构的一个关键参数。过渡金属之所以表现出独特的催化活性,很大程度上是因为它们拥有未完全填充的、空间延展适中的d轨道。
d带中心被普遍定义为d电子投影态密度(PDOS)的质心(centroid)或一阶矩。态密度n(E)描述了在能量E附近单位能量间隔内的量子态数量。而投影态密度则是将总态密度投影到特定原子(如表面某个金属原子)的d轨道上,从而专门反映该原子d电子的能态分布情况。

其中:nd(E)是d电子的投影态密度。E是能量,通常相对于费米能级(EF)进行度量。分母∫nd(E)dE是d带的总电子态数,分子∫End(E)dE是d电子态密度按能量加权后的积分。

图6 Co/Co6Mo6C2及相关样品的d带中心。DOI:10.1002/adfm.202519088
在实际应用中,特别是在研究与吸附相关的成键过程时,研究者更关心已占据的d电子态。因此,积分上限有时会被设为费米能级EF以计算已占据d带的中心:

这里的分母代表d带的填充数。在大多数文献讨论中,除非特别说明,d带中心通常指的是相对于费米能级计算得到的质心值,其数值的正负和大小直接反映了d电子态能量的平均高低。一个更负的值意味着εd带的能量重心更低,离费米能级更远。
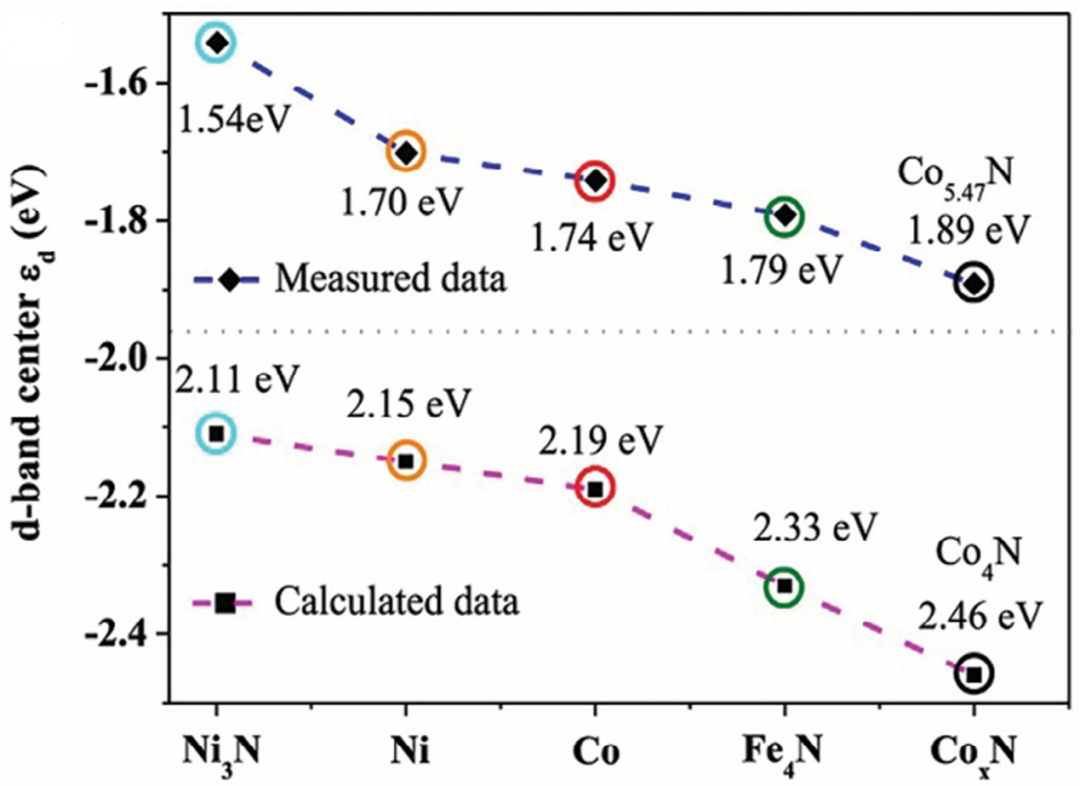
图7 d带中心的测量值和计算值。DOI:10.1002/advs.202308040
d带中心物理内涵与意义
d带中心提供了一个极其简洁的标量,用以概括复杂的d带电子结构(一个包含多个峰和谷的函数nd(E))的整体能量位置。它之所以如此重要,是因为它与过渡金属表面和吸附物种之间化学键的强度直接相关,这是d带中心理论的核心。
反映能量平均值:从物理上看,εd代表了d电子的平均能量。这个平均能量的高低,反映了d电子的反应活性。
作为反应描述符:一个较高的d带中心(即εd值更接近于0 eV,或为更小的负数)意味着d电子的平均能量较高,这些电子更不稳定或更有可能参与成键,因此通常对应着更强的表面反应性和更强的吸附能力。
反之,一个较低的d带中心(εd值为更大的负数)则意味着d电子平均能量较低,d能带整体更稳定,与吸附物的相互作用也更弱。

图8 d带中心和催化活性的比较。DOI:10.1021/jp303276z

全局性与局域性
费米能级是一个全局性的、热力学性质的参数,决定了电子转移的宏观趋势。对于一块宏观的、导电的材料,在平衡状态下,其内部所有位置的费米能级(电子化学势)都是统一的。
d带中心则是一个局域(local)性的、与能带结构相关的参数,精细地刻画了过渡金属d电子的反应活性。它被定义在单个原子或一类等效的原子上。它描述的是特定原子周围d电子云的能量分布重心。
在同一个材料中,表面原子的d带中心通常与体相原子的d带中心不同;合金中不同元素原子的d带中心也不同。

图9 不同Pt层对应不同的d带中心。DOI:10.1007/s40042-023-00934-3

图10 不同PtNi(hkl)表面取向对应不同的d带中心。DOI:10.14279/depositonce-7828
体系属性与轨道属性
费米能级是整个电子体系的属性,它由体系的总电子数和所有可用的量子态(包括s,p,d,f等所有轨道)共同决定。改变总电子数(如充电或掺杂)会直接改变费米能级。
d带中心是d轨道的内禀属性,主要由d轨道的能量、形状和d-d轨道间的相互作用(决定d带宽度)决定。虽然它也受s,p带杂化的影响,但其核心是d电子的行为。


图11 费米能级(EF)、d带中心是筛选过渡金属催化剂的重要参数。DOI:10.1038/s41524-022-00922-4
相对位置的核心意义
费米能级和d带中心并非两个孤立的参数。在催化科学的语境中,它们的相对位置,即εd-EF,才是真正具有决定性意义的物理量。这个差值量化了d电子态的能量重心与体系中可用于化学反应的最高能量电子之间的距离。
当d带中心升高(更接近)时:d电子的平均能量更高。这导致与吸附物轨道杂化时,形成的成键态和反键态能量都相应升高。由于反键态整体上移,其被费米能级以下的电子填充的比例就会减少。
这种“反键态填充的减少”效应,虽然听起来像是会增强化学键,但实际上,主导作用是d带与吸附物轨道的耦合强度增强,导致了更强的吸附。因此,d带中心越靠近费米能级,化学吸附越强。

图12 d带中心理论DOI:10.1016/B978-0-443-19256-2.00006-5
当d带中心降低(远离)时:d电子的平均能量更低,更稳定。与吸附物轨道的能量差增大,耦合减弱。这导致杂化效应减弱,形成的化学键也相应减弱。因此,d带中心越远离费米能级,化学吸附越弱。

图13 纯金属表面的压缩和拉伸应变改变d带中心与费米能级距离。DOI:10.14279/depositonce-7828
相互影响的复杂机制
改变一个参数往往会引起另一个参数的变化,但这种变化并非简单的线性关系。假设有一个刚性带模型,如果d带形状不变,向体系中添加电子,如通过合金化引入电子数更多的元素,这些电子会填充到费米能级以上的最低未占据态,从而导致费米能级升高。
然而,现实远比刚性带模型复杂。当合金化或施加应变时,不仅电子数会变,原子间的相互作用、轨道杂化程度都会改变。这会导致d带本身的形状(宽度、对称性)和位置(中心)都发生变化。d带的展宽或收缩会重新分布态密度,进而影响为了容纳总电子数所需要的费米能级的位置。