

Bader电荷是一种基于电子密度的原子电荷分析方法,由Richard Bader提出,广泛应用于化学、材料科学和计算化学领域。
该方法通过将分子或晶体的电子密度划分为原子的“Bader体积”,从而计算每个原子的净电荷。这种方法不仅能够直观地展示原子间的电荷分布,还能用于分析化学键的性质、电荷转移机制以及材料的电子结构特性。
以下华算科技将从Bader电荷的理论基础、计算方法、应用场景以及相关研究进展等方面进行详细阐述。



Bader电荷的理论基础
Bader电荷的核心思想是基于Richard Bader的“原子定义”理论,该理论认为,一个原子的电荷可以通过其在电子密度场中的“零通量面”来划分。
零通量面是指电子密度在垂直于表面方向上达到最小值的二维表面,通常在分子系统中,原子之间的电荷密度达到最小值,这是分离原子的理想位置。通过这种方式,Bader电荷能够更准确地反映分子内部电子密度的变化,从而揭示原子间的相互作用和电荷转移机制。
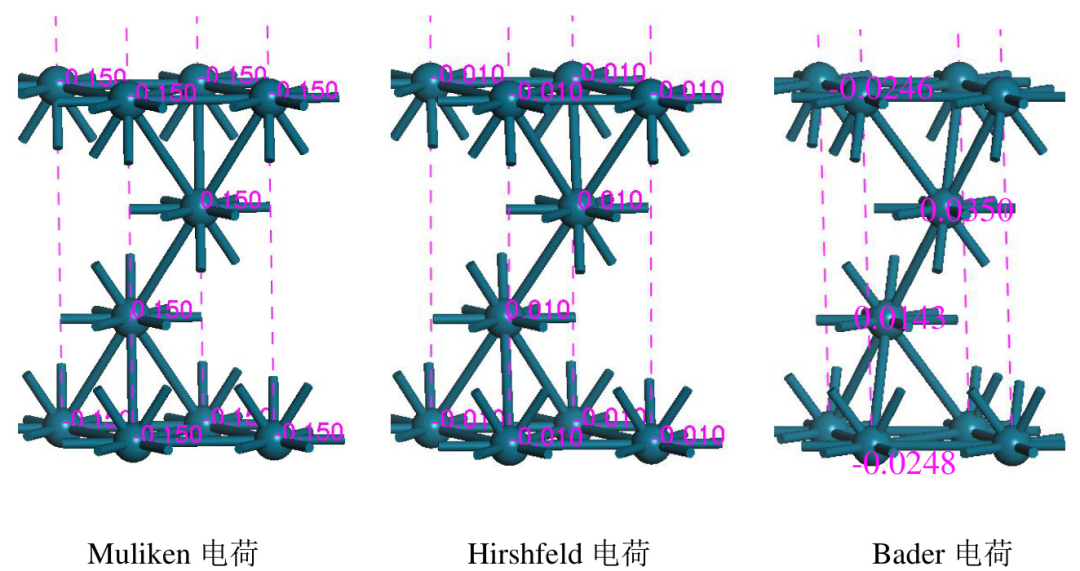
Bader电荷的计算依赖于电子密度的数值模拟,通常使用密度泛函理论(DFT)计算电子密度,并通过特定的算法(如Bader分解算法)将电子密度划分为原子的Bader体积。
这种方法的优势在于,它完全基于实验数据,不需要引入任何经验参数,因此具有较高的可靠性和普适性。
Bader电荷的计算方法
Bader电荷的计算通常涉及以下几个步骤:
结构优化:首先需要对目标分子或晶体进行结构优化,确保其处于最低能量状态。这一步骤可以通过第一性原理计算软件(如VASP、Quantum ESPRESSO等)完成。
单点能计算:在优化后的结构上进行单点能计算,生成电子密度文件(如CHGCAR文件)。在计算过程中,需要设置LAECHG=.TRUE.,以确保生成的电子密度文件包含足够的信息用于后续的Bader电荷分析。
生成电子密度总和文件:使用chgsum.pl脚本将AECCAR0和AECCAR2文件合并为CHGCAR-sum文件,这是Bader电荷计算的关键步骤之一。
运行Bader电荷分析程序:下载并安装Bader电荷分析程序(通常可以从理论计算中心获取),并运行bader命令,指定输入文件为CHGCAR,参考文件为CHGCAR-sum。程序将生成ACF.dat、BCF.dat和AtomVolumes.dat等输出文件,其中ACF.dat包含了每个原子的Bader电荷值。
结果解读:通过getCharges脚本提取ACF.dat中的Bader电荷值,并结合其他计算结果(如电荷密度分布、键长、键角等)进行分析。
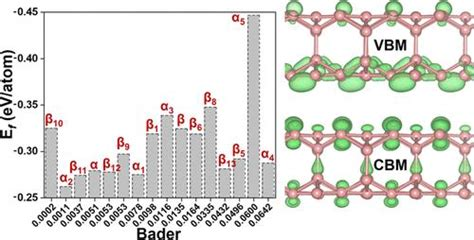
Bader电荷的应用场景
Bader电荷在多个领域具有广泛的应用,主要包括以下几个方面:
电催化反应研究
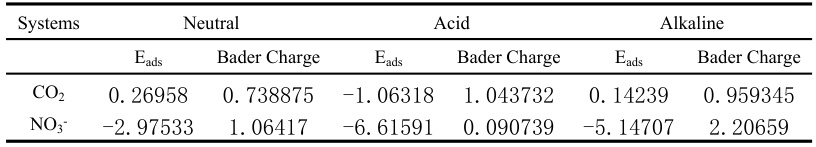
在电催化反应中,Bader电荷可以用于分析反应物和产物之间的电荷转移机制。例如,在CO2还原反应中,Bader电荷可以揭示CO2分子在催化剂表面的吸附行为及其电荷分布。
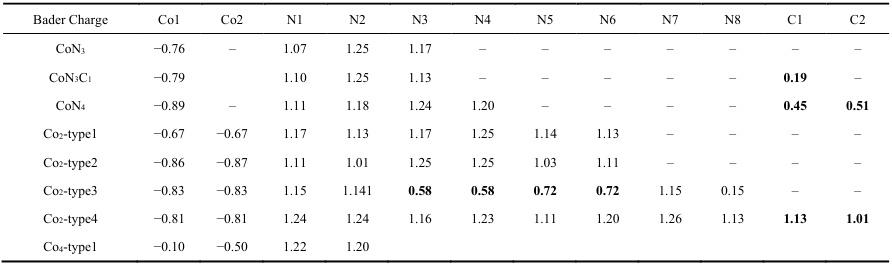
研究表明,不同pH条件下的Bader电荷值可以显著影响CO2和NO3⁻在高熵硼化物(HEBs)表面的吸附行为。此外,在析氧反应(OER)中,Bader电荷可以用于分析钴基氮碳催化剂的构效关系,揭示不同Co-N-C结构中的电荷分布。
界面反应研究
在界面反应研究中,Bader电荷可以用于分析界面处的电荷转移和化学键形成。例如,在MoO3/Si界面反应中,Bader电荷可以揭示钼掺杂非晶氧化硅层的形成机制。
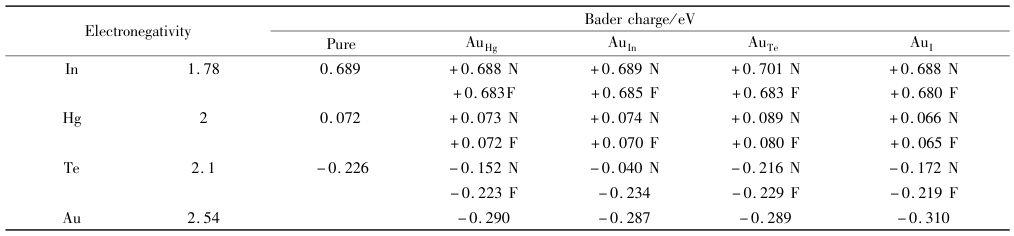
研究表明,Sb和Cu掺杂可以降低Cl原子在Fe(100)表面的吸附倾向,抑制Cl原子和Fe之间的电荷交换。此外,在Au掺杂Hg3In2Te6的成键机制研究中,Bader电荷可以揭示掺杂元素对电子结构的影响。
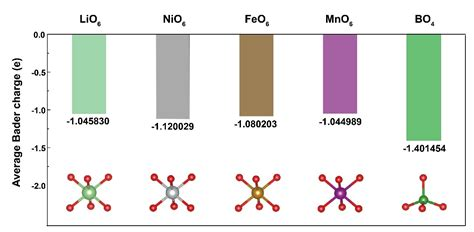
材料设计与性能预测
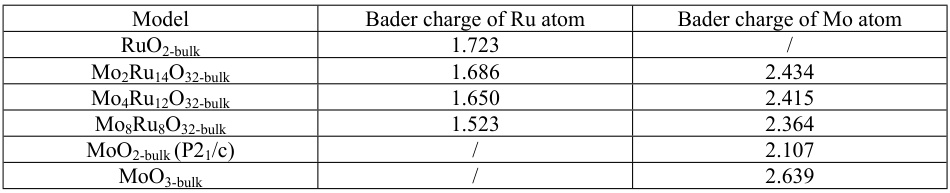
在材料设计中,Bader电荷可以用于预测和优化材料的性能。例如,在石墨烯表面的羰基模型中,Bader电荷可以揭示不同官能团的电荷分布,从而指导材料的改性设计。此外,在Ru-Mo合金的水氧化反应中,Bader电荷可以揭示Ru和Mo之间的电子相互作用,从而优化合金的催化性能。
化学键分析
Bader电荷可以用于分析化学键的性质,例如键的极性、键的强度和键的类型。通过比较不同基组下的Bader电荷值,可以揭示基组对电荷分布的影响。
例如,在甲烷(CH4)中,Bader电荷和Mulliken电荷的比较表明,Bader电荷能够更准确地反映碳原子和氢原子之间的电荷分布。
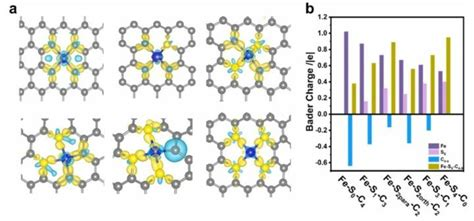
此外,Bader电荷还可以用于分析不同元素之间的电荷转移,例如在Cl原子吸附在Fe(100)表面时,Bader电荷可以揭示Cl原子和Fe原子之间的电荷交换。
Bader电荷的计算工具与软件
Bader电荷的计算通常依赖于特定的软件和工具,主要包括以下几种:
VASP:VASP(Vienna ab initio simulation package)是一种广泛使用的第一性原理计算软件,支持Bader电荷的计算。在VASP中,可以通过设置LAECHG=.TRUE.和LCHARG=.TRUE.来生成电子密度文件,并通过chgsum.pl脚本和bader程序进行Bader电荷分析。
Bader程序:Bader程序是一个专门用于Bader电荷分析的工具,可以从理论计算中心下载。该程序可以读取VASP生成的CHGCAR文件,并通过零通量面算法计算每个原子的Bader电荷值。
Gaussian:Gaussian是一种常用的量子化学计算软件,支持Bader电荷的计算。在Gaussian中,可以通过设置IOp(3/33=1)来生成电子密度文件,并通过bader程序进行Bader电荷分析。
CASTEP:CASTEP是一种基于密度泛函理论的材料模拟软件,支持Bader电荷的计算。在CASTEP中,可以通过设置lcharg=.true.来生成电子密度文件,并通过bader程序进行Bader电荷分析。