

说明:在量子化学和凝聚态物理中,自旋多重度(Spin Multiplicity) 是一个描述体系电子总自旋角动量的关键物理量。
它深刻影响着原子、分子及固态物质的电子结构、磁学性质和化学反应活性。在基于密度泛函理论(DFT)等的第一性原理计算中,正确设定和理解自旋多重度是获得可靠计算结果的先决条件。本文从自旋多重度的定义、第一性原理中的运用和实际案例三个方面进行了详细介绍。
什么是自旋多重度?
自旋多重度的定义式为2S+1,其中S是体系的总自旋量子数。总自旋量子数S是体系中所有电子自旋量子数 (si=1/2) 的矢量和的绝对值。
一个更直观的计算方法是,S等于体系中未成对电子数n的一半,即S=n/2。因此,自旋多重度可以表示为n+1。
单重态 (Singlet):当体系中没有未成对电子时 (n=0),S=0,自旋多重度为1。此时所有电子自旋都已配对。
双重态 (Doublet):当体系中有1个未成对电子时 (n=1),S=1/2,自旋多重度为2。自由基通常处于双重态。
三重态 (Triplet):当体系中有2个未成对电子时 (n=2),S=1,自旋多重度为3。根据洪德规则,氧分子的基态即为三重态。
更高多重度:依此类推,有3个未成对电子时为四重态 (n=3, S=3/2),以此类推。
自旋多重度决定了在磁场中电子能级的分裂情况,是解读光谱实验数据和理解材料磁性的基础。
第一性原理计算中的自旋多重度
第一性原理计算,特别是基于DFT的方法,已经成为预测和解释材料性质的强大工具。在进行这类计算时,必须为所研究的体系指定一个初始的自旋多重度。
这个设定直接决定了计算中所求解的电子波函数和能量,对于开壳层体系(含有未成对电子的体系)尤为重要。
关键作用与应用:
确定基态和激发态:对于一个给定的原子或分子,不同自旋多重度的态对应着不同的电子排布和能量。计算不同自旋多重度下的体系总能量,可以帮助确定其磁性基态。
例如,通过比较单重态和三重态的能量,可以判断一个双自由基体系的基态是反铁磁性耦合还是铁磁性耦合。
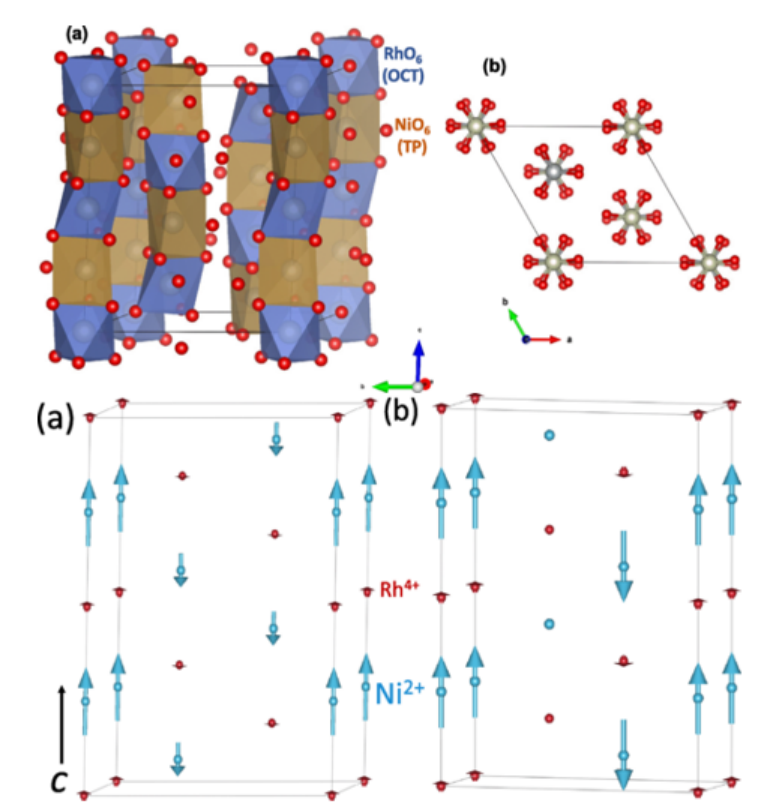

DOI: 10.1103/PhysRevB.110.224415
预测磁学性质:自旋多重度与体系的总磁矩直接相关。在第一性原理计算中,通过设定不同的自旋构型(如铁磁、反铁磁、亚铁磁),可以计算材料的磁矩、磁各向异性以及交换作用参数,这对于自旋电子学材料的设计至关重要。
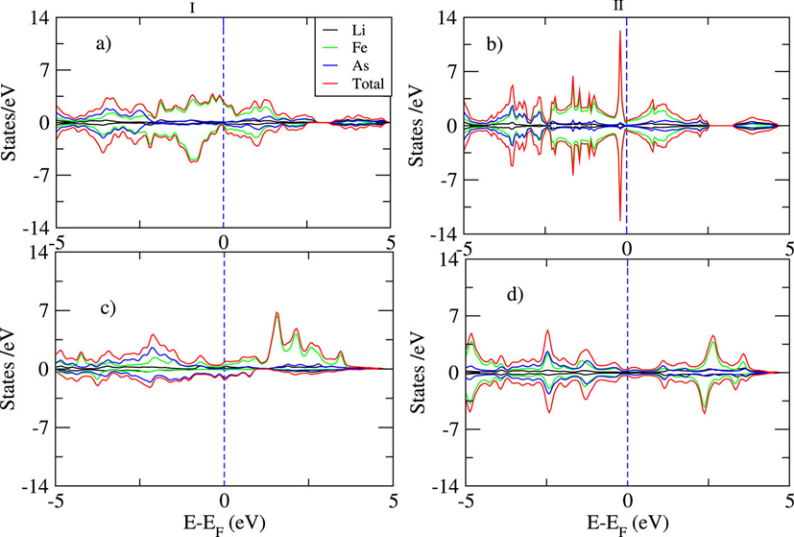
DOI: 10.1063/5.0269638
理解化学反应机理:化学反应过程中常常伴随着化学键的断裂与生成,这会导致电子自旋状态的改变。因此,追踪反应路径上自旋多重度的变化对于理解反应机理,特别是涉及自由基或过渡金属的催化反应,是不可或缺的。
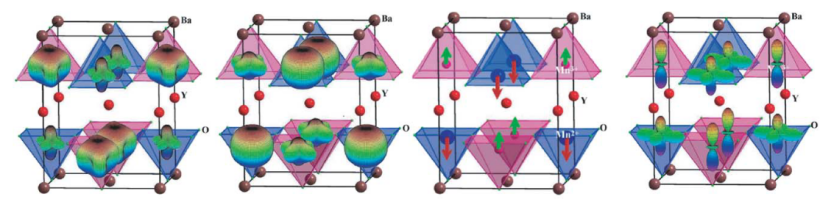
DOI: 10.1016/S0022-4596(03)00215-9
自旋多重度的实际运用
缺陷物理与电荷态
在半导体材料中,点缺陷(如空位、间隙原子)的性质与其电荷态和自旋态密切相关。
一篇发表在Physical Review B上的研究“Spin multiplicity and charge state of a silicon vacancy (TV2a) in 4H-SiC determined by pulsed ENDOR” 就结合了第一性原理计算和实验手段,精确确定了碳化硅中硅空位的自旋多重度为四重态 (S=3/2),从而纠正了之前对其电荷态的错误认识。
这表明,精确的自旋多重度计算对于理解缺陷的物理化学性质至关重要。
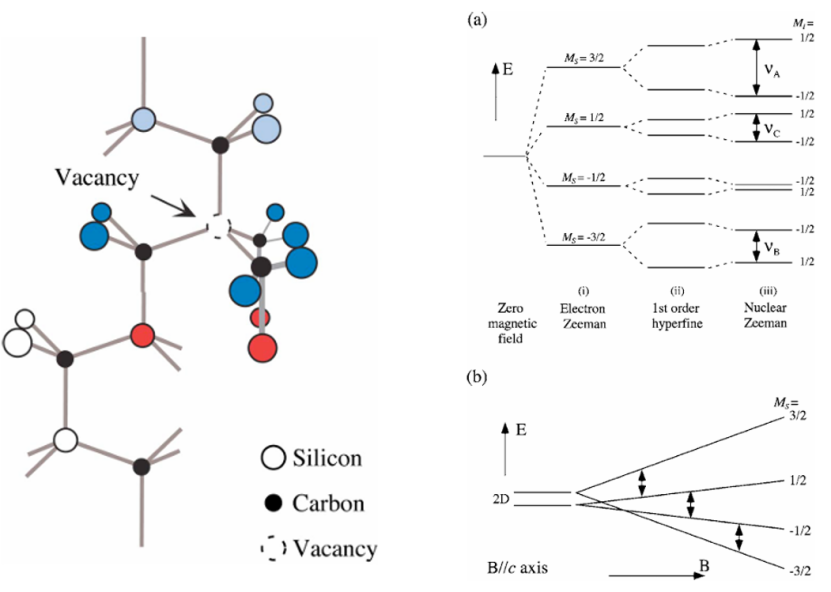
DOI: 10.1103/PhysRevB.72.235208
团簇与纳米材料物理
在纳米尺度下,团簇的性质往往与其尺寸和电子结构紧密相关。一篇发表在ResearchGate上的论文“The Influence of Spin Multiplicity on the Melting of Na55(+)” 通过第一性原理分子动力学模拟,研究了自旋多重度对钠团簇熔化行为的影响。
研究发现,高自旋态的基态构型赋予了团簇额外的结构稳定性。这说明在纳米材料的理论研究中,必须考虑自旋多重度的影响。
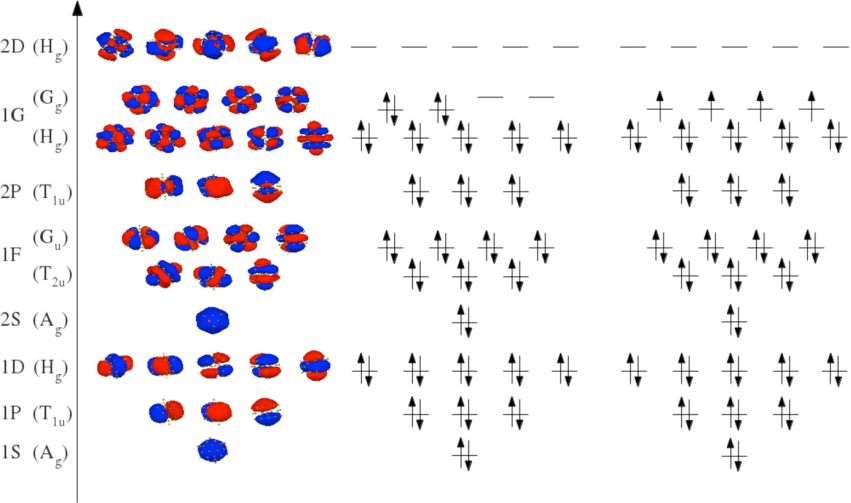
DOI: 10.1021/acs.jpclett.5b01983
复杂化学反应的挑战
传统DFT计算需要在计算前手动指定自旋多重度。然而,对于许多复杂的化学反应,其反应路径上自旋态可能会发生改变,事先确定其变化非常困难。
ACS Omega上的一篇论文 “Computational Analysis of Chemical Reactions Using a Variational Quantum Eigensolver Algorithm without Specifying Spin Multiplicity” 探讨了利用量子计算来解决这一难题。
量子计算机能够制备不同自旋多重度的叠加态,原则上只需一次计算就能找到体系的电子基态,极大地简化了对复杂反应的研究。这代表了该领域未来一个重要的发展方向。
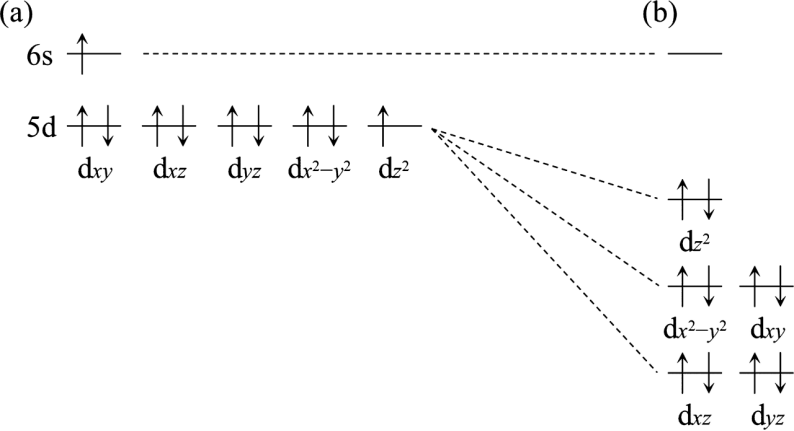
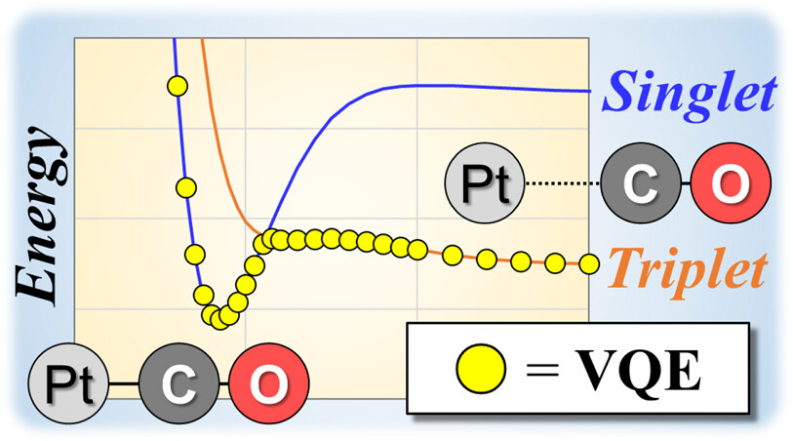
DOI: 10.1021/acsomega.3c01875
自旋电子学与磁性材料设计
国内外的许多综述性文章,如材料科学与工程学院网站上刊登的《综述:自旋电子学材料的第一性原理设计》以及《物理学报》上的《自旋涨落与非常规超导配对》,都强调了通过第一性原理计算调控电子自旋以设计新型功能材料的重要性。
这些研究的核心之一就是精确计算和预测材料在不同自旋构型下的稳定性和电子结构。
总结
自旋多重度是连接微观电子行为与宏观材料性质的关键桥梁。在第一性原理计算中,它不仅是一个必须设定的计算参数,更是理解体系电子结构、磁性、反应活性的核心。
随着计算方法的发展和新算法(如量子计算)的出现,对自旋多重度的研究将继续深入,为材料科学、化学和物理学等领域带来新的见解和突破。