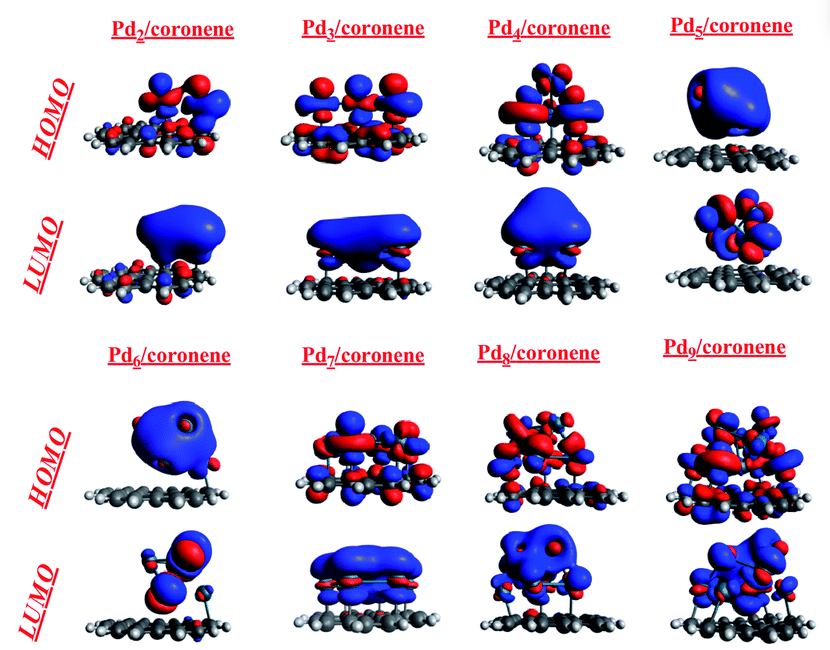
其中,最高占据分子轨道(Highest Occupied Molecular Orbital,HOMO)和最低空分子轨道(Lowest Unoccupied Molecular Orbital,LUMO)的能量与空间分布,成为决定分子光学、电学及化学行为的重要指标。
HOMO与LUMO之间的能量差,通常被称为分子能隙(HOMO-LUMO gap),在本质上对应了分子由基态跃迁到激发态的最低能量要求。这一物理量不仅决定了分子的稳定性和反应活性,也在很大程度上影响了分子对光的吸收与发射、电荷转移效率以及电子结构调控方式。
随着量子化学计算方法与光谱学实验技术的快速发展,HOMO-LUMO理论已逐渐从最初的有机分子化学研究,拓展到凝聚态物理、光电子学、分子电子学以及催化反应动力学等众多领域。
理论基础与分析方法
HOMO-LUMO理论起源于分子轨道理论。分子轨道可由原子轨道线性组合(LCAO-MO)获得,其中HOMO代表电子在基态分布中最高能量的轨道,而LUMO则是最低能量的未占据轨道。
由于化学反应往往发生在电子密度最活跃的区域,HOMO作为电子供体,LUMO作为电子受体,便成为研究分子反应性的重要指标。
从理论上看,HOMO能级反映了分子的电离势,而LUMO能级则对应电子亲和能,两者之间的差值即为分子能隙。这一能隙越大,分子通常越稳定,化学反应性越低;反之,能隙越小,分子越容易参与电子跃迁与反应。
在计算方法上,密度泛函理论(DFT)是最常见的工具。通过不同的泛函选择(如B3LYP、PBE0等),可以较为精确地获得分子HOMO和LUMO的能量与波函数分布。此外,含时密度泛函理论(TD-DFT)则能够进一步分析分子的电子激发态性质,预测吸收光谱和电荷转移过程。
实验方面,紫外–可见光谱(UV-Vis)、电化学循环伏安法(CV)、扫描隧道显微镜(STM)等技术都可提供HOMO-LUMO相关的间接或直接证据。例如,通过CV实验测得的氧化还原电位,可以转化为HOMO与LUMO能级,从而辅助解释实验中的光电或催化现象。
HOMO-LUMO分析并非只限于能量差大小的比较,更重要的是空间分布的考察。若HOMO轨道主要分布在分子的某一部分,而LUMO轨道则位于另一部分,则分子在电子激发过程中容易发生分子内电荷转移(ICT)。这种特性对于光敏染料、分子光伏器件、以及单分子电子器件的设计尤为重要。
因此,对HOMO与LUMO的全面分析,既需要能量数值的精确计算,也需要轨道分布图的可视化与解释,从而为理解与预测分子体系的性能提供可靠依据。
在分子光学与光电领域的应用
在光学与光电领域,HOMO-LUMO能隙被广泛用于解释和预测材料的吸收、发射与电荷传输特性。分子对光的吸收通常来源于HOMO至LUMO的电子跃迁,因此能隙的大小决定了吸收光谱的起始位置。
例如,能隙较小的分子能够在可见光甚至近红外区吸收,从而适合于光电探测器与太阳能电池等应用。相反,能隙较大的分子则主要在紫外区吸收,常用于光学涂层与紫外探测材料。
在光致发光与电致发光过程中,HOMO与LUMO的空间分布影响着激子的形成与复合效率。如果两者分布在分子的不同部分,容易形成电荷转移态,从而实现发射波长的可调控与激子寿命的延长。
这一机制在有机发光二极管(OLED)中尤为重要,因为其效率依赖于单重态与三重态激发态的能级差,而这一能级差正是由HOMO与LUMO的分布所决定。在非线性光学材料中,HOMO-LUMO的对称性和能量差决定了分子的极化率,从而影响其在光通信与光调制领域的应用潜力。
因此,HOMO-LUMO能级不仅是解释分子光学特性的理论依据,更是材料设计与器件性能优化的重要指导工具。通过调控分子结构以改变HOMO-LUMO能隙与分布,研究者能够实现对光学性质的精确设计,从而推动有机光电技术的发展。
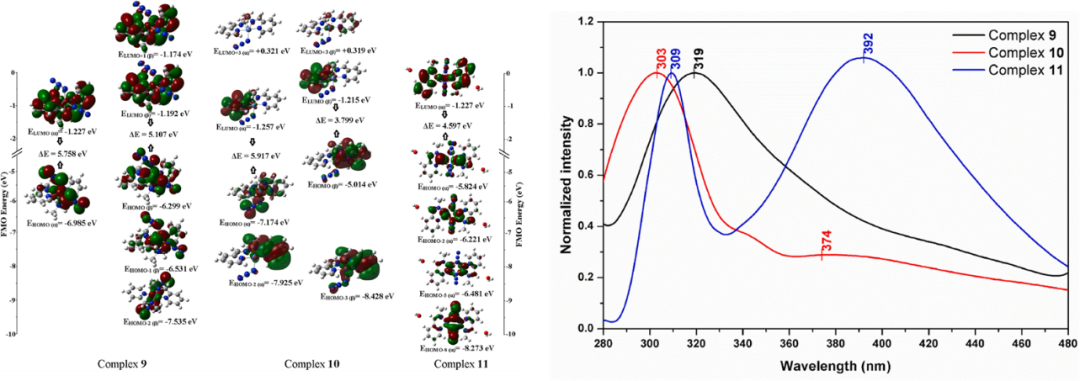

在化学反应与催化中的应用
在化学反应机理研究中,HOMO与LUMO的相互作用是反应发生的根本动力。根据前线分子轨道理论,化学反应往往由一个反应物的HOMO与另一个反应物的LUMO之间的能级匹配所驱动。当两者能级差较小且空间重叠显著时,电子跃迁更为容易,反应势垒显著降低。
这种分析方式不仅适用于简单的取代反应,也适用于复杂的协同反应,如Diels–Alder反应。通过比较反应物HOMO-LUMO能隙,可以预测反应的相对速率与选择性。
在催化领域,尤其是过渡金属催化与光催化中,HOMO-LUMO理论具有重要意义。金属催化剂的d轨道能级常常接近反应物的HOMO或LUMO,从而促进电子转移并降低反应活化能。
HOMO与LUMO的匹配不仅解释了催化反应的选择性,还揭示了配体与金属中心的电子效应在催化中的作用机制。在光催化和电催化体系中,催化剂的HOMO与LUMO能级位置决定了电荷分离与转移的效率。
例如,催化剂LUMO能级过低时,能够有效接受电子,从而促进还原反应;HOMO能级位置则影响其失电子能力,从而决定氧化反应的进行。
由此可见,HOMO-LUMO理论不仅在有机反应机理中提供了直观解释框架,也在催化剂设计与优化中发挥着核心作用。
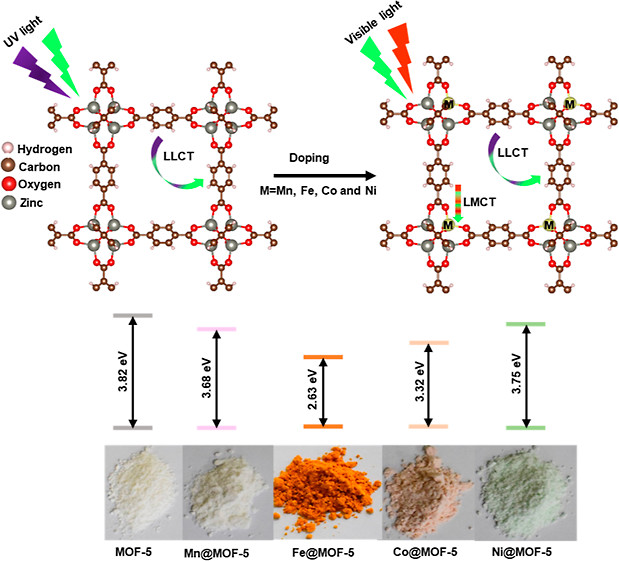
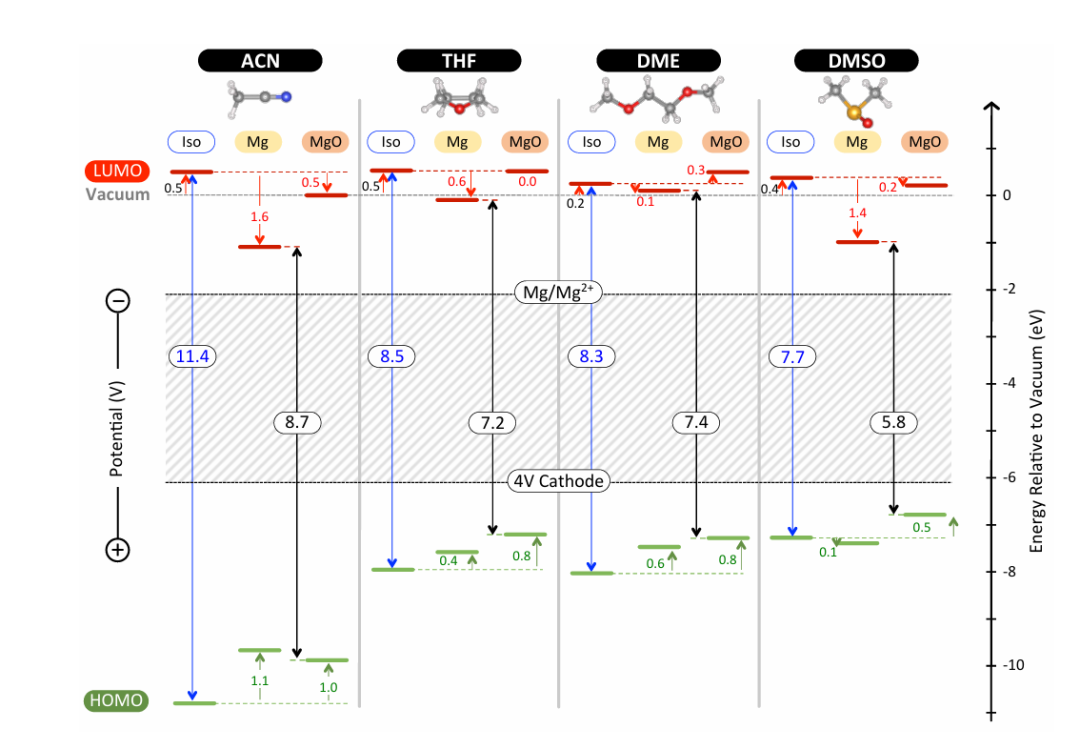
在电池电解液中的应用
在电化学储能体系,尤其是锂离子电池、钠离子电池、固态电池以及有机电池的发展中,电解液的分子结构与电子特性直接决定了电池的安全性、循环寿命和能量密度。电解液分子的HOMO与LUMO能级分析,为理解其电化学稳定窗口提供了核心理论依据。
电解液分子的氧化稳定性通常由其HOMO能级决定,当HOMO能级过高时,分子容易失去电子而在高电位下被氧化分解;而其LUMO能级位置则与还原稳定性密切相关,当LUMO能级过低时,电解液分子在低电位下极易接受电子发生还原分解。
因此,HOMO与LUMO共同界定了电解液的电化学稳定区间,这一窗口必须覆盖电池正负极的工作电位,才能保证整个电化学循环的可逆性与长期稳定运行。这一分析方法不仅应用于传统的碳酸酯类电解液设计,也被广泛推广到离子液体、电解质添加剂以及新型固态电解质的开发中。
从分子设计的角度来看,通过调控电解液分子的化学结构,可以有效调节其HOMO-LUMO能级。例如,在碳酸酯体系中,通过在分子骨架上引入氟原子或电子吸引基团,可以有效降低分子HOMO能级,从而增强其抗氧化能力,使其适用于高电压正极材料,如富镍三元正极或高电压尖晶石型材料。
在钠离子电池与多价离子电池中,电解液分子往往需要同时具备较高的还原稳定性,以避免在低电位下发生副反应,因此通过LUMO能级调控来优化其电化学性能成为研究热点。
另一方面,在固态电解质体系中,聚合物电解液与无机电解质的HOMO-LUMO分析有助于理解其界面稳定性,尤其是在与锂金属负极接触时,LUMO能级过低的材料容易发生不可控还原反应,从而导致界面阻抗上升甚至电池失效。
因此,HOMO-LUMO分析在设计电解液分子、调控界面反应机理、拓展电池电压窗口方面具有不可替代的理论价值。随着高能量密度电池的快速发展,电解液的稳定性成为制约其实际应用的关键瓶颈。HOMO-LUMO理论结合DFT计算,已成为筛选新型电解液分子与添加剂的重要手段。
通过计算得到的HOMO-LUMO能隙不仅能够预测分子的本征稳定性,还能够与实验中的循环伏安法、X射线光电子能谱等结果相互印证,从而加速电解液分子的优化与筛选。
此外,近年来的研究表明,电解液分子在界面上发生的成膜过程(SEI膜与CEI膜的形成)与其HOMO-LUMO分布密切相关。
当LUMO能级接近锂金属的费米能级时,分子更易被还原形成固体电解质界面膜(SEI),而HOMO能级过高的分子在高电压下则更可能贡献于正极电解质界面膜(CEI)的形成。这种基于HOMO-LUMO的能级匹配分析,正在成为构建稳定、高效电极/电解液界面的重要设计原则。
在凝聚态物理与材料科学中的应用
在凝聚态物理中,HOMO-LUMO概念被拓展为能带理论中的价带顶(VBM)与导带底(CBM)。对于有机半导体材料而言,分子HOMO-LUMO能级在凝聚态相互作用下形成价带与导带,进而决定材料的导电性与光学带隙。
因此,HOMO-LUMO的研究不仅局限于单分子层面,还扩展到材料能带结构的理解。在二维材料、有机–无机杂化钙钛矿以及分子晶体中,HOMO-LUMO调控策略成为决定其电输运性质与光电性能的关键手段。
在纳米材料与低维体系中,HOMO-LUMO能隙的量子限域效应尤为显著。随着材料尺寸减小,能隙会随之增大,从而改变材料的光吸收与发射特性。例如,量子点材料的光致发光波长正是由其HOMO-LUMO能隙决定的,因此通过调控纳米晶粒径即可实现对发光颜色的精确控制。
在有机分子电子学中,单分子的HOMO与LUMO能级决定了其作为分子二极管、分子晶体管的工作机制。这类应用将传统分子轨道理论与现代纳米电子学紧密结合,为未来分子尺度器件的发展奠定了基础。
此外,在功能材料设计中,HOMO-LUMO的调控也是性能优化的重要途径。通过引入不同取代基、调节分子共轭结构,研究者能够有效调控能隙大小,使材料在导电性、光学性质、化学稳定性等方面达到特定需求。
这一策略在导电聚合物、光敏涂层、防腐材料等领域均有广泛应用。因此,HOMO-LUMO理论作为连接分子物理与材料科学的桥梁,正在推动凝聚态物理与应用科学的深度融合。