

说明:COOP与COHP是理解材料中化学键性质与催化性能的核心电子结构分析方法。COOP从电子占据角度揭示成键行为,COHP则从能量贡献角度分析键稳定性。二者互补,能够精准识别催化反应中活性位与中间体之间的成键强弱,帮助理解掺杂、缺陷等结构调控对催化性能的影响。
在HER、OER、CO₂RR等典型电催化反应中,COOP/COHP结合DFT计算、PDOS、电荷分析等手段,为催化剂筛选与反应路径优化提供了理论依据。其逐步标准化的应用方式,正在推动电催化机制研究走向可视化、定量化和系统化。


COOP与COHP
晶体轨道重叠布居(COOP, Crystal Orbital Overlap Population)和晶体轨道哈密顿布居(COHP, Crystal Orbital Hamilton Population)是分析固体材料电子结构中化学键性质的重要工具。
在理论上,两者都通过对能带结构的电子波函数进行分解,提供键合(bonding)与反键合(antibonding)信息,但方式有所不同。COOP方法将材料的电子态密度(DOS)按轨道间的重叠布居加权,从而显示特定原子对之间在各能量下的键重叠程度。
COHP方法则以哈密顿矩阵元为权重,将能带能量在原子对之间划分,从能量角度评估键强度。简单而言,COOP按电子数划分键类型,而COHP按能量划分键强弱。
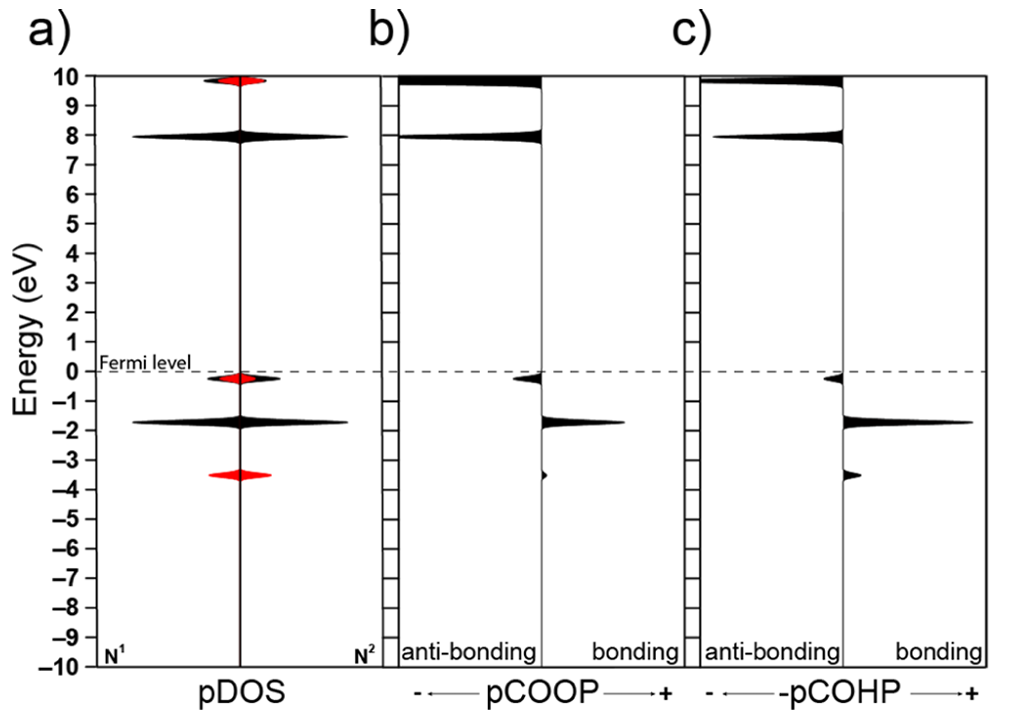
DOI:10.1021/acs.jpcc.8b08934
具体来说,COOP曲线在某一原子对的能量–电子数空间描述该对键的性质:曲线取正值表示键合作用,负值表示反键合作用。
这是因为正的轨道重叠布居意味着两个原子轨道同相叠加形成稳定键,而负值对应异相叠加形成不稳定的反键轨道。相比之下,COHP通过将每一态的能量按原子对的哈密顿矩阵耦合元来加权,刻画键结合对体系能量的贡献。
由于稳定键的形成会降低体系能量,在COHP中键合作用对应负的哈密顿矩阵元(降低能量),反键作用对应正的矩阵元。科研人员常取-COHP来作图,使得键合态呈现为正向峰,反键合态为负向峰,与COOP曲线的直观习惯一致。
需要注意的是,COOP与-COHP曲线虽然外观相似但并非完全等价;COOP侧重电子占据数,COHP侧重能量贡献,各自提供的信息侧重点不同。
两种分析的积分值含义也不同:将COOP曲线积分到费米能级,可得到对应原子对的总重叠电子数,类似于传统Mulliken布居分析中的键级概念。将COHP曲线积分(取负号后)则得到一个能量值,可视作该键对体系稳定性的相对度量。
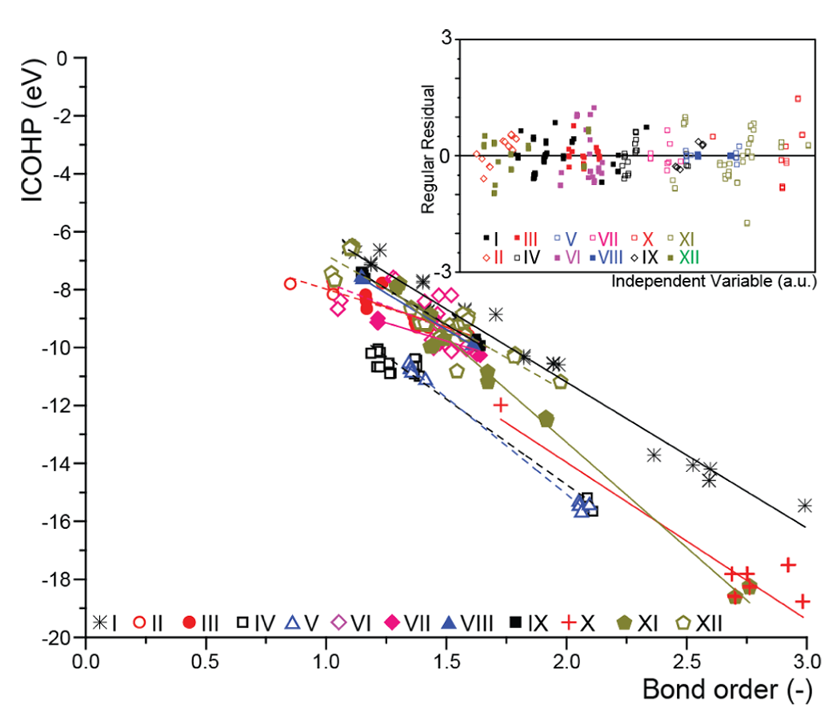
这一积分值常称为集成COHP(ICOHP);更负的ICOHP(或更大的-ICOHP)一般表示键合作用更强,即键能更高。然而需谨慎的是,ICOHP并不严格等同于真实化学键能,因为COHP只划分了一电子能量而未直接对应体系总能量。
尽管如此,在不同材料和键的比较中,ICOHP仍被证明与实际键强度和键级趋势高度相关。例如,有研究发现COHP得到的键能指标与基于电子密度的键级呈良好线性关系。
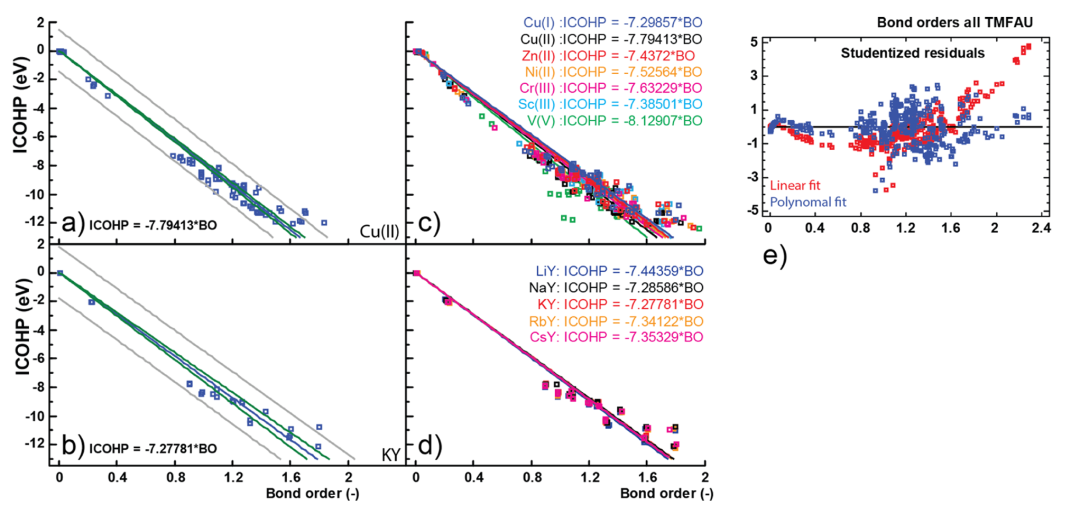
DOI:10.1021/acs.jpcc.8b08934
综上,COOP和COHP分别从电子占据和能量贡献两方面为研究者提供了定量刻画键(反)成键特征的手段,其中COOP反映轨道重叠与键序,COHP反映键的能量和强度。
COOP/COHP的应用
电催化过程(如HER、OER、ORR、CO₂RR等)涉及催化剂表面原子与反应物/中间体之间复杂的成键作用。COOP和COHP分析已成为顶尖电催化研究中理解这些作用、揭示活性机制的重要工具。
研究者通过第一性原理计算(DFT)获取材料的电子结构,然后利用COOP/COHP考察关键键合的能态分布和强弱,从而解释催化性能差异。
在HER(析氢反应)研究中,COHP常用于分析金属催化剂与吸附氢(H*)之间的键:例如在CoP等过渡金属磷化物中,通过COHP曲线可以比较掺杂前后金属-H键的键合强度变化,从而理解掺杂调控HER活性的原理。
有报道显示,向CoP中引入适量异金属(如Cr)能改变Co–H键的COHP分布,减少反键成分并增强成键峰,意味着H*吸附更适宜,从而提升碱性HER速率。
类似地,在镍氮化物Ni₃N表面负载CeO₂的电催化体系中,COHP分析帮助证明了引入氧化物促进了产氢中间体OH的脱附:COHP曲线显示改性后Ni–O键在费米能级以下出现更多反键成分,使OH键合更易断裂。
这种“填充反键态促进中间体脱附”的现象与实验观察的性能提升一致,直接将电子结构变化与催化行为相关联。
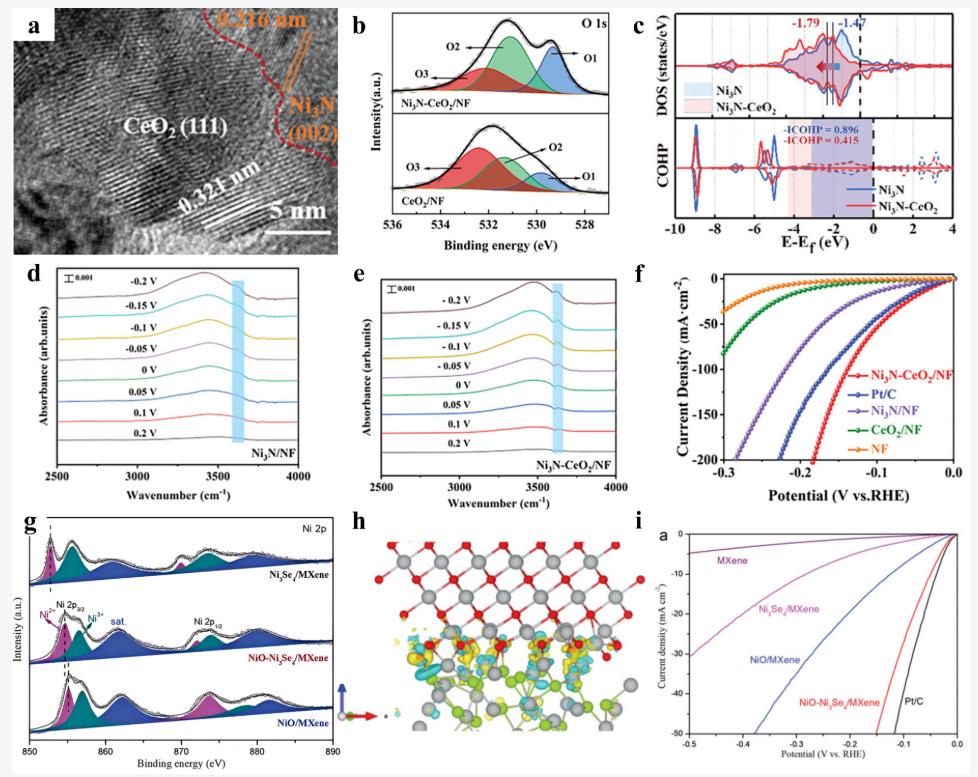
DOI:10.3390/nano15141106
在OER和ORR等氧相关反应中,COHP/COOP更是被广泛用于研究金属–氧键和中间体键合。例如,对于单原子催化剂上的ORR(氧还原)过程,研究者通过投影COHP(pCOHP)分析吸附中间体(O、OH、OOH)与催化剂间的键合情况,揭示打破传统线性标度关系的原因。
在一系列过渡金属掺杂磷烯(TM–Ph)的计算研究中,pCOHP显示某些催化剂(如Ni、Cu、Ag基的单原子催化剂)对O中间体的成键峰和反键峰展开宽度变化较大,导致不同金属对O和OH键强的比值各异,进而打破了OH与OOH吸附能之间的线性关系。
这种非线性使部分单原子催化剂同时拥有较弱的O吸附(避免毒化表面)和适度的OH吸附,从而优化了ORR的反应路径。
pCOHP分析进一步发现:这些催化剂在价带顶端成键态占比略高于反键态,意味着金属–氧键处于“不过强也不过弱”的最优键合状态,既稳定中间体又不过度钝化活性位。
这样的电子结构解释了为何某些材料在ORR中表现出优异活性,同时避免了活性位被氧中间体毒化的问题。总的来说,COHP/COOP曲线帮助识别了催化活性位上键合过强或过弱的情形,为优化催化剂提供了直观指引。
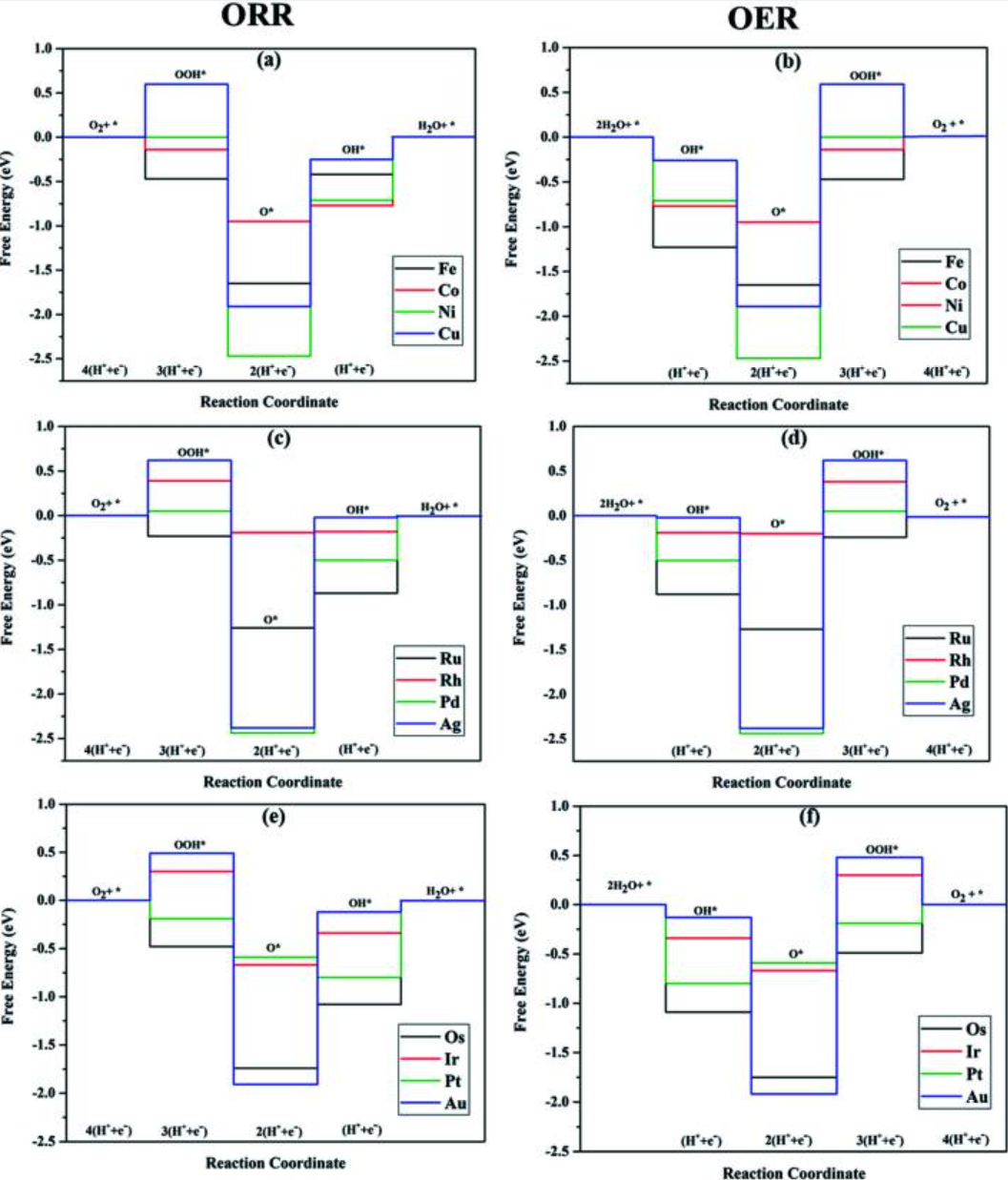
DOI:10.1039/d0na00209g
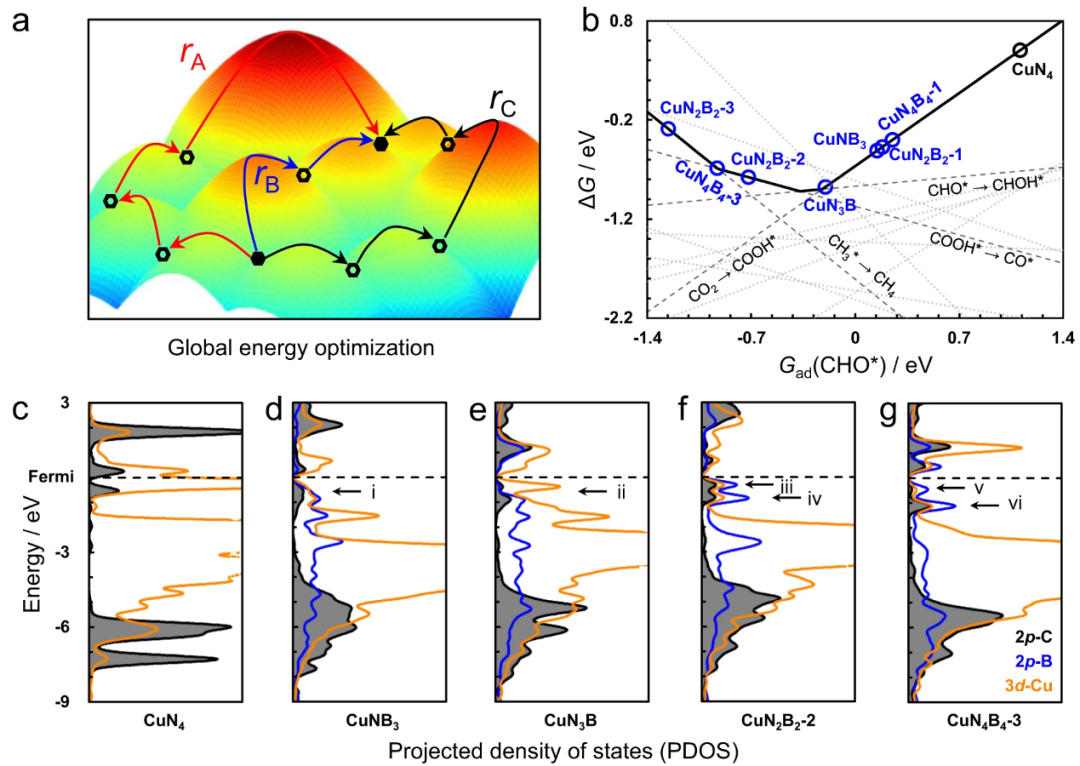
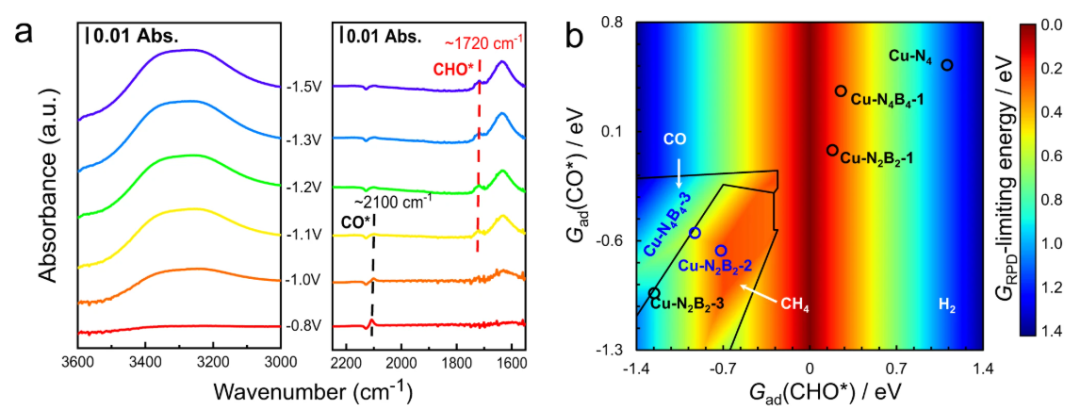
在CO₂电还原(CO₂RR)等复杂多电子反应中,COHP同样发挥了独特作用。例如,在Cu基单原子催化剂将CO₂选择性转化为CH₄的研究中,COHP用于解析关键中间体CHO的吸附键合。
结果表明,在未掺杂的Cu–N₄位点上,CHO主要通过C–Cu键吸附,但COHP曲线显示该C–Cu键的反键轨道部分被占据(抗键峰有人电子),削弱了CHO的稳定性。
而引入电负性更低的B原子取代部分N(形成Cu–N₃B₁等配位环境)后,CHO同时与Cu和B发生键合:COHP分析揭示新形成的C–B键强烈成键(无明显反键占据),其键合强度显著高于原先的C–Cu键。
换言之,B掺杂增强了关键中间体与活性位的相互作用。结合电子态分析,研究者发现掺杂后CHO的2p轨道与Cu 3d、B 2p轨道产生明显杂化,共振峰位置下移,这与COHP得到的更强成键作用相符。
这一系列计算结果成功解释了实验上观察到的选择性提升:B调控使Cu单原子位对CO和CHO等中间体具有更强的吸附,从而抑制了CO中间产物过早解吸,转而促进了深度质子化生成CH₄。
类似方法还被应用于分析CO₂RR中C–C偶联反应的过渡态,在不同表面上比较CO–CO键形成时金属–C键的COHP值,以判断哪些结构有利于降低C–C成键能垒。
例如在铜催化CO₂RR生成C₂产物的研究中,COHP揭示了四配位的“方形”低配位位点能同时提供4个Cu–C键,使*OC–CO过渡态中的C–Cu键强度(-ICOHP值)显著高于三配位或双配位位点,从而稳定过渡态并降低反应能垒。这解释了缺陷位点(如阶梯/拐角上形成方形Cu簇)比理想晶面更易生成C₂产物的现象。
DOI:10.1038/s41467-023-39048-6
综上,在各类电催化顶刊研究中,COOP/COHP分析已成为剖析关键键合、关联电子结构与催化性能的常规手段。从氢吸附到氧中间体、碳中间体,研究者借助这些工具找出了活性位上“最佳键合强度”的判据:过强会造成位点钝化,过弱则活化不足。
使用趋势与评价
随着第一性原理计算在电催化领域的普及,COOP/COHP分析在近年顶刊文献中出现频率大幅提高,已成为主流的键合分析工具之一。这一下游趋势得益于计算工具的发展(如LOBSTER程序使得可以在平面波DFT结果上投影计算COHP),以及研究者对深入理解催化机制的需求。
COOP/COHP能够提供传统仅看吸附能或d带中心等参数所无法直接给出的键性质细节,对于揭示催化活性位电子结构具有独特价值。
在分析催化活性位点时,COHP曲线清晰展示了活性位与反应物/中间体之间哪些能级是成键的、哪些是反键的,以及它们相对费米能级的位置。这使科学家能够判定某键是否过于强烈以致占据了反键轨道(从而不稳定)或过于薄弱(成键轨道几乎为空)。
通过对照不同材料或不同表面,COHP提供了一种图谱式的比较手段:哪一种调整(如掺杂、缺陷引入、相结构改变)能够减少不利的反键填充或增强成键作用,从而优化催化性能。
因此,近年的研究中COHP常被用来评估电子结构调控策略的有效性,如证明某掺杂确实降低了键合占据的反键态、某构型确实增加了键合的共价程度等。
DOI:10.1038/s41467-023-39048-6
在调控电子结构和预测活性方面,不少文献已将COHP/ICOHP等量化参数作为活性预测指标。
例如,对于氧还原反应,有工作提出以M–O键的ICOHP值作为描述符来筛选单原子催化剂,发现适中的ICOHP对应最佳ORR活性,并通过大量计算验证了这一趋势。
这说明COHP方法不只是事后分析工具,还具备一定的预测功能:通过理论计算COHP,可以在催化剂合成前预判其关键键合是否理想,从而加速材料设计。当然,也有文献对COHP提出谨慎意见,指出ICOHP等指标需要结合其它证据综合判断,不能机械地将其当作绝对的键能替代。
例如,由于COHP忽略了电子相关能等因素,它可能高估金属键在过渡态稳定化中的作用,因此在涉及反应能垒时通常和差分电荷、能量分析一起讨论。
DOI:10.1021/acs.jpcc.8b08934
COOP/COHP已被广泛认可为理解电催化机制的强有力工具:它以化学直观的方式将电子结构与催化性能联系起来。许多顶级研究者在发表的评述中都强调了COHP分析的重要性,认为其对于在原子水平上设计更优催化剂不可或缺。
可以预见,随着研究的深入,COOP和COHP方法将在电催化领域获得更加标准化和普及化的应用,同时可能出现与实验谱学结合的新用法,进一步提高对催化剂工作机制的认识。
COHP在顶级电催化研究中的应用
“Manipulating local coordination of copper single atom catalyst enables efficient CO₂-to-CH₄ conversion” 研究旨在提高铜基单原子催化剂在CO₂还原生成甲烷过程中的选择性,通过在Cu–N₄配位结构中引入硼(形成Cu–N₂B₂配位)来调节Cu的电子结构。
COHP分析在这项研究中发挥了关键作用,用于揭示为什么引入B会增强对中间体的吸附,从而提高CH₄产率。
论文首先通过DFT计算比较了纯Cu–N₄和B掺杂的Cu–N₂B₂位点对关键中间体CHO的吸附。COHP结果显示:在纯Cu–N₄上,CHO通过C原子与Cu键合,但该C–Cu键的反键态在费米能级以下被部分占据,即COHP曲线在价带顶附近出现明显负值峰。
这表示CHO与Cu键合时存在未稳定因素,可能导致中间体结合不够牢固。而在B掺杂的Cu–N₂B₂位点,CHO的C原子同时与Cu和邻近的B发生键合。
COHP分析清楚地表明:C–B键的成键轨道几乎完全占据且没有显著反键填充,其峰强度远大于C–Cu键。换句话说,掺杂B使CHO获得了一个更强且稳定的额外锚定键,从而大大增强了CHO在活性位的吸附。
研究者还结合PDOS发现,CHO*在掺B后的体系中2p轨道与Cu 3d/B 2p轨道形成了强烈杂化(在价带出现新的共振峰),印证了电子相互作用的增强。
综合COHP和PDOS结论,作者提出:在Cu–N₄中引入更弱电负性的B,会向Cu提供额外电子密度,提升Cu对碳中间体的亲和力,同时由于B与中间体形成强共价键,避免了C–Cu键过弱导致的中间体提早脱附。
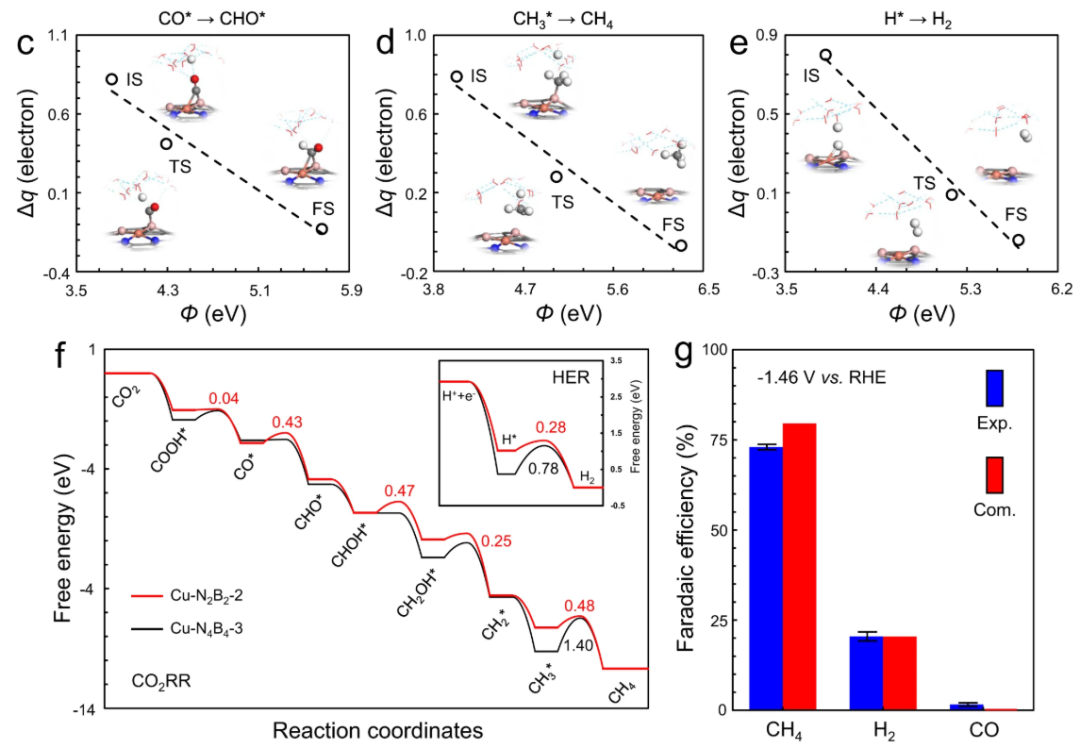
电化学测试结果也支持这一机制:B掺杂的单原子Cu催化剂表现出大幅提高的CH₄法拉第效率和电流密度,而未掺杂的对比样品主要产物是CO(说明未掺杂时CO中间体倾向脱附成为副产物)。
COHP分析在该论文中直接证明了掺杂调控的微观作用,使作者能够自洽地将电子结构改变与宏观性能提升联系起来。
通过这个案例可以看到,COHP曲线所揭示的反键态占据与否成为判断催化剂优劣的关键:Cu–N₄存在反键电子导致结合不理想,而Cu–N₂B₂消除了该缺陷,因而活性提高。
论文结论中特别强调了COHP方法的重要性,指出“PDOS与COHP分析清晰地表明B的引入促进了关键中间体的吸附,从而提升了反应活性”。
这一表述凸显了COHP在理解催化剂作用机理上的贡献:没有它,电子结构的细微差别(如反键轨道部分占据)很难被发现,更难以和催化效果对应起来。
DOI:10.1038/s41467-023-39048-6
总的来说,COOP/COHP分析在电催化领域为科研人员提供了一双“电子之眼”,可以透视活性位的化学键,帮助回答“为什么这个催化剂更活跃”的核心问题。
展望未来,COOP和COHP方法将继续在电催化机理研究中扮演不可或缺的角色,帮助我们设计出更高效、更有针对性的催化材料,从基础理论层面加速清洁能源技术的发展。
总结
COOP(晶体轨道重叠布居)与COHP(晶体轨道哈密顿布居)是第一性原理计算中广泛应用的电子结构分析工具,专用于揭示固体材料中化学键的性质及其在催化反应中的作用机理。
两者都通过对能带结构的处理,揭示原子间轨道重叠与成键特征,但本质不同:COOP以轨道重叠布居为基础,强调电子占据行为,用正负值区分成键与反键;COHP则从哈密顿矩阵出发,将成键对体系总能量的影响量化,常取负值绘图(-COHP)以统一成键/反键的表达方式。
积分形式的COOP和COHP分别对应于“电子占据数”与“键稳定化能量”,为电子结构研究提供了多角度解析。特别在电催化领域,COOP/COHP已成为分析催化剂与反应中间体之间成键强度、识别反应活性位点、评估掺杂/缺陷效应等问题的重要手段。
与态密度(PDOS)、电荷密度差、Bader电荷等方法结合使用时,COHP/COOP能够有效刻画“电子如何参与键形成、键断裂”,揭示吸附强弱变化背后的微观原因,是连接电子结构与催化性能的关键桥梁。
其应用在HER、OER、CO₂RR等反应体系中已得到大量验证,特别是在单原子催化剂和异质结催化剂研究中尤为重要。未来,COOP与COHP有望进一步与机器学习、材料筛选数据库结合,推动电子结构驱动的催化剂设计进入高通量、可预测的新阶段。