本文详细探讨了电催化硝酸盐还原反应(NO3RR)的三种主要反应路径及其特点。路径一涉及多步脱氧和加氢过程,路径二通过直接还原生成中间产物,路径三以羟胺(NH₂OH)为关键中间体。
通过DFT计算和自由能分析,对比了不同路径的复杂程度、副产物生成及热力学优势,揭示了路径三在某些条件下具有较低反应能垒的优势。此外,结合具体案例(如Nb掺杂异质结、CuCo(111)合金及单原子催化剂Ti/V/Nb/g-C₃N₄),分析了不同催化剂对反应路径选择性和活性的影响。
研究表明,催化剂的设计(如金属中心特性、吸附强度)和反应条件调控是提高NO3RR效率与氨选择性的关键。这些发现为高效氨合成催化剂的开发提供了理论依据和实验指导。


NO3RR反应路径概述
NO3RR是一个复杂的电化学反应过程,涉及多个电子转移步骤和中间产物的生成。根据DFT计算和实验研究,NO3RR主要存在三种反应路径,每种路径都有其独特的反应步骤和特点。
路径一:
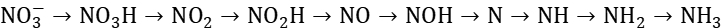
此路径最为复杂,涉及多个中间产物的逐步脱氧和加氢过程。在这个过程中, 首先被质子化形成NO3H,然后逐步失去氧原子并加氢,最终生成NH3。每一步反应都伴随着电子的转移,整个过程共涉及8个电子的转移。
首先被质子化形成NO3H,然后逐步失去氧原子并加氢,最终生成NH3。每一步反应都伴随着电子的转移,整个过程共涉及8个电子的转移。
路径二:
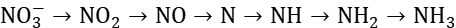
该路径相对直接,通过较少的中间步骤实现硝酸盐到氨的转化。直接还原为NO2,然后依次经过NO、N等中间产物,最终生成NH3,同样涉及8个电子的转移。
路径三:
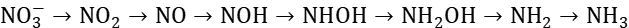
此路径以羟胺(NH2OH)为关键中间产物,通过特定的反应步骤实现硝酸盐的还原。先转化为NO2,再经过NO、NOH等中间产物生成NH2OH,最后NH2OH进一步加氢生成NH3,也是8电子转移过程。
不同路径对比分析
三种反应路径在复杂程度、副产物生成和热力学优势等方面存在显著差异。
路径一最为复杂,涉及的中间步骤最多,这使得反应过程中产生副产物的可能性增加,如可能产生NO和N2O等副产物。这些副产物的生成不仅会降低氨的选择性,还可能对环境造成负面影响。
路径二相对直接,但由于其反应过程中N原子的存在形式较为简单,使得N2的生成概率增加,同样不利于氨的选择性合成。
路径三以NH2OH为关键中间产物,虽然减少了N2的生成,但NH2OH的稳定性较差,需要在反应过程中进行有效的调控,否则可能会分解或参与其他副反应。
从热力学角度来看,路径三在一定条件下具有优势。根据DFT计算,路径三的某些关键步骤的反应能垒相对较低,使得反应更容易朝着生成氨的方向进行。
这是因为路径三中的中间产物NH2OH具有特殊的电子结构,使得其在加氢过程中更容易发生反应,从而降低了反应的整体能垒。
此外,路径三的反应过程中,各中间产物之间的转化相对较为顺畅,没有明显的能量瓶颈,这也使得该路径在热力学上更有利于氨的生成。
然而,实际反应中路径的选择还受到多种因素的影响,如催化剂的种类、反应条件(如温度、压力、pH值等)以及反应物和产物的浓度等。在不同的条件下,三种路径的反应速率和选择性可能会发生变化,因此需要综合考虑各种因素,以实现NO3RR的高效、高选择性转化。
NO3RR案例分析
路径1
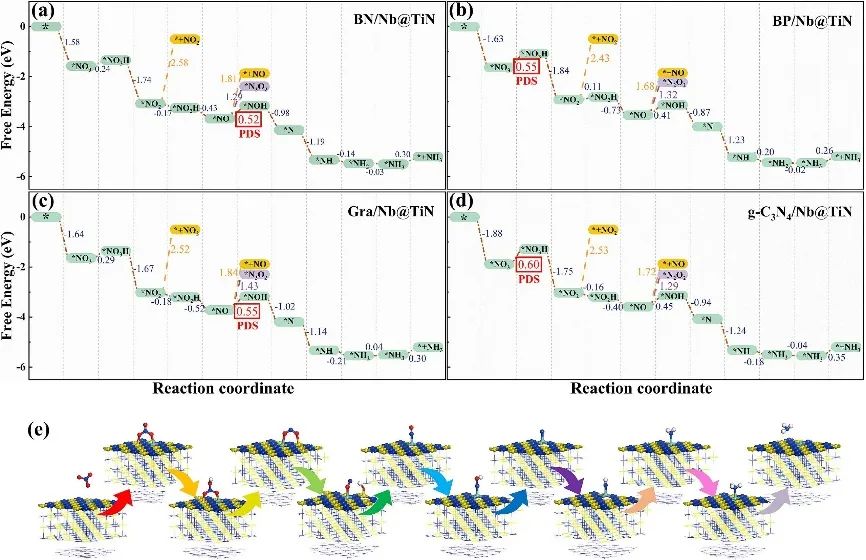
下图(a-d)展示了BN/Nb@TiN、BP/Nb@TiN、Gra/Nb@TiN和g-C₃N₄/Nb@TiN四种Nb掺杂异质结的NO₃RR反应自由能图谱,横轴为反应坐标,纵轴为吉布斯自由能(单位为 eV),同时对比了生成副产物NO₂和NO的路径能量变化。
从图中可知,四种体系的决速步骤(PDS)均出现在氢化反应阶段,即*NO+H⁺→NOH或NO₃+H⁺→*NO₃H,这与文献报道一致。
其中,BN/Nb@TiN的PDS自由能变化为0.52 eV,BP/Nb@TiN和Gra/Nb@TiN均为 0.55 eV,g-C₃N₄/Nb@TiN为 0.60 eV,这些数值低于或接近此前报道的V-锚定石墨炔(0.63 eV)、V/g-CN(0.64 eV)等VB族单原子催化剂,表明其高催化活性。
从反应路径细节来看,在硝酸盐还原为氨的过程中,N-O键的断裂是关键步骤。以BN/Nb@TiN为例(图 e),初始步骤*NO₃+H⁺→NO₃H 的吉布斯自由能升高,表明质子攻击导致N-O键弱化,随后NO₂→NO₂H 步骤的ΔG为-0.17 eV,系统能量降低,反应自发进行,伴随一个N-O键断裂,暴露出N原子以进一步氢化。
在后续N→NH₃的氢化步骤中(包括N+H⁺→*NH、*NH+H⁺→*NH₂、*NH₂+H⁺→NH₃),除Gra/Nb@TiN的NH→*NH₂步骤需克服0.04 eV能垒外,其余步骤的吉布斯自由能均为负值,确保氨的顺利生成。
综上,Nb掺杂的2D/TiN异质结通过低能垒的氢化步骤、高效的N-O键活化及抑制副反应路径,展现出优异的NO₃RR催化性能,其中BN/Nb@TiN、BP/Nb@TiN和Gra/Nb@TiN在能量效率和选择性方面表现尤为突出。
DOI: 10.1016/j.seppur.2025.131524
路径2
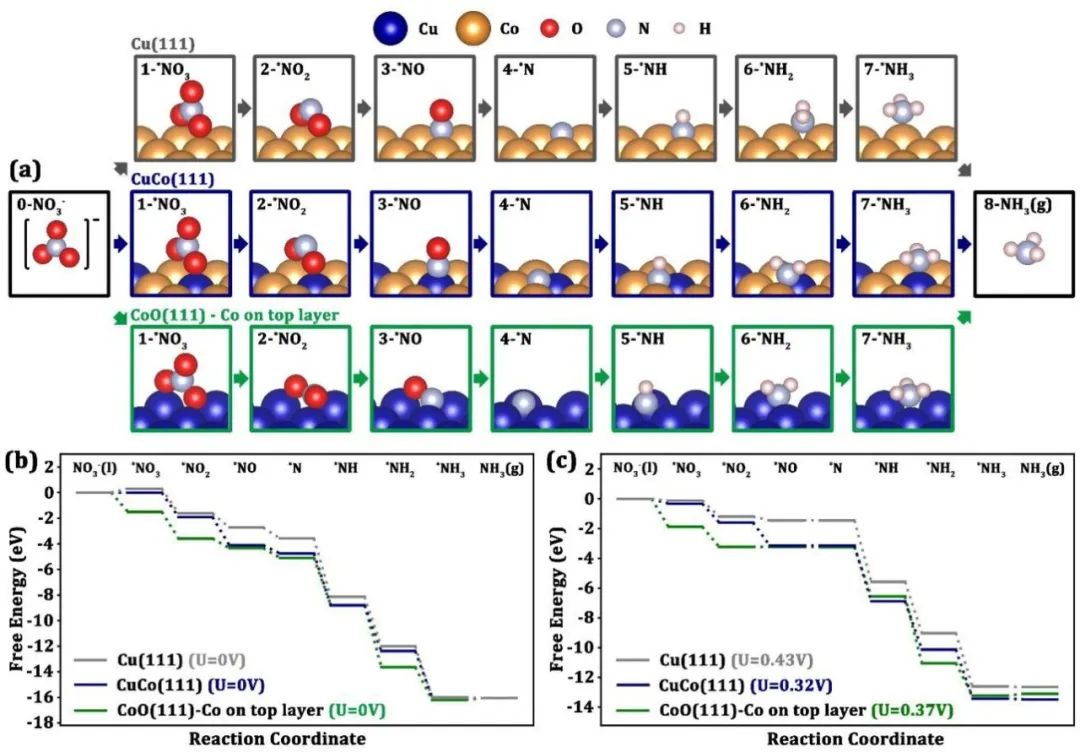
下图展示了Cu(111)、CuCo(111)和CoO(111)三种表面模型在硝酸盐电还原反应(NO₃RR)中的自由能变化路径,揭示了不同材料对反应机理和活性的影响。
对于Cu(111)表面,NO₃RR的起始步骤是硝酸盐吸附,随后通过一系列加氢和N-O键断裂步骤生成中间体,最终形成NH₃并脱附。整个反应路径包含八个步骤,涉及八电子转移过程。
Cu(111)的潜在决定步骤(PDS)是从NO到N的加氢步骤,自由能变化为0.43 eV,对应的极限电位为0.43 V。在CuCo(111)表面上,反应路径与Cu(111)类似,但PDS的自由能变化降低至0.32 eV(极限电位0.32 V),表明CuCo合金的引入提高了催化活性。
CoO(111)表面的反应路径同样遵循八电子转移机制,但其PDS的自由能变化为0.37 eV(极限电位0.37 V),介于Cu(111)和CuCo(111)之间。
值得注意的是,尽管Cu(111)在理论上表现出较高的NO₃RR活性,但在实际电化学测试中,CuCo-CoO复合电催化剂的性能优于纯Cu/C催化剂,这主要归因于CuCo和CoO对竞争反应氢析出反应(HER)的抑制能力更强。
具体而言,CuCo(111)和CoO(111)表面在较低电位下(分别为0.08 V和-0.45 V)即可抑制HER,而Cu(111)需要更高的电位(0.12 V),从而在复合催化剂中为NO₃RR提供了更多的活性位点。
此外,DFT计算还显示,CoO(111)表面的氢吸附能力极低,进一步抑制了HER,提高了氨的选择性。
在稳定性方面,CuCo(111)和CoO(111)的凝聚能(分别为-3.63 eV/atom和-3.87 eV/atom)比Cu(111)(-3.27 eV/atom)更负,表明复合催化剂具有更高的热力学稳定性,这与实验中CuCo-CoO在48小时内保持40%初始活性的结果一致。
总体而言,通过自由能路径分析,明确了Cu(111)、CuCo(111)和CoO(111)在NO₃RR中的反应机制和活性差异,其中CuCo(111)表现出最低的极限电位,而CoO(111)对HER的抑制能力最强。
这些结果与全文的实验数据相吻合,解释了CuCo-CoO复合催化剂在氨产率(12.63 mmol·h⁻¹·cm⁻²)和法拉第效率(96.68%)上的优越性能,以及其显著增强的稳定性。
DOI: 10.1016/j.apsusc.2023.157118
路径3
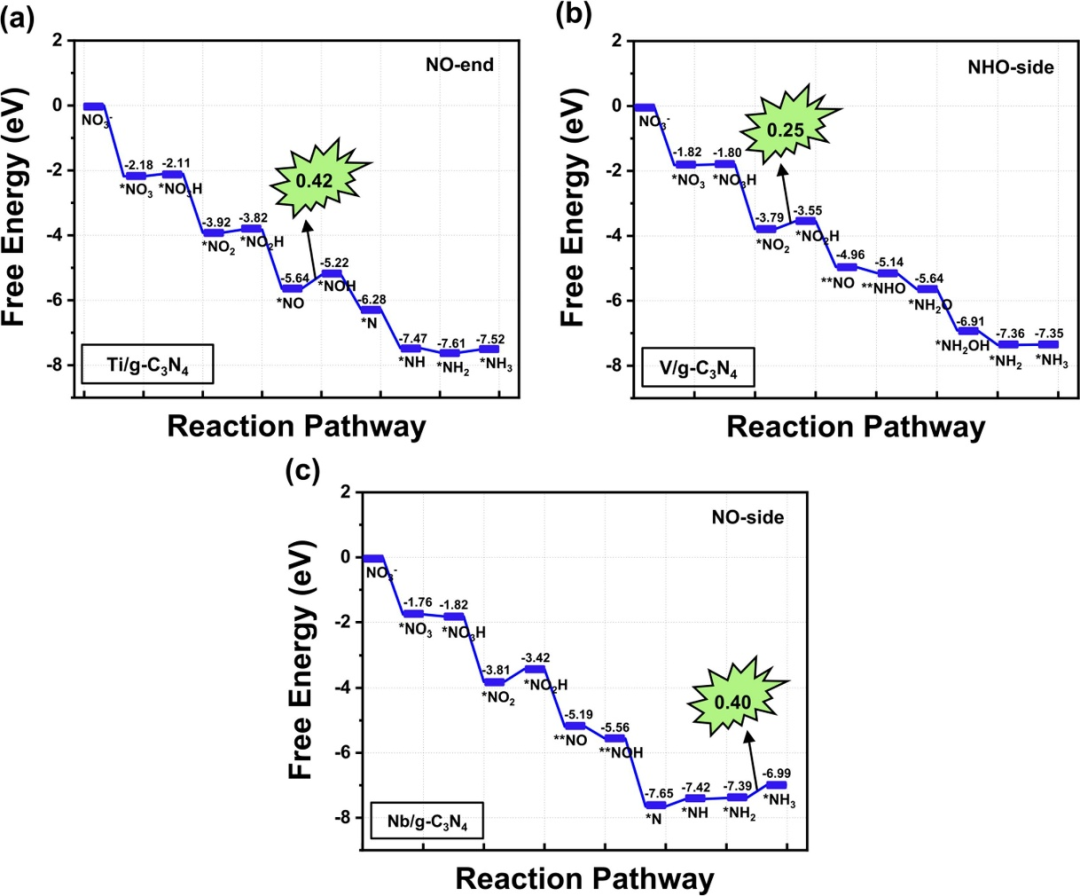
下图展示了Ti/g-C₃N₄、V/g-C₃N₄和Nb/g-C₃N₄三种单原子催化剂在硝酸盐电还原反应(NO₃RR)中的自由能变化路径,揭示了不同金属中心对反应机理和活性的影响。
对于Ti/g-C₃N₄,反应起始于NO₃⁻通过η²-O构型吸附在Ti位点上,吸附自由能为-2.18 eV,表现出较强的吸附能力。
随后,NO₃⁻经过两步加氢(NO₃ → NO₂H和NO₂H → NO)生成NO中间体,其中NO → *NOH步骤的自由能变化为0.42 eV,成为潜在决定步骤(PDS),对应的极限电位为-0.42 V。
在V/g-C₃N₄中,NO₃⁻的吸附自由能为-1.82 eV,反应路径倾向于NHO-side机制,*NO₂ → *NO₂H步骤的自由能变化为0.25 eV(PDS),极限电位为-0.25 V,表明V/g-C₃N₄具有更高的催化活性。
Nb/g-C₃N₄的NO₃⁻吸附自由能为-1.76 eV,反应遵循NO-side机制,*NH₂ → *NH₃步骤的自由能变化为0.40 eV(PDS),极限电位为-0.40 V。三种催化剂的反应路径均显示,NO₃⁻的吸附和活化是反应的关键初始步骤,而后续加氢步骤的自由能变化决定了整体反应的效率。
此外,还表明,Ti/g-C₃N₄和Nb/g-C₃N₄的PDS分别出现在*NO → NOH和NH₂ → NH₃步骤,而V/g-C₃N₄的PDS出现在NO₂ → *NO₂H步骤,反映了不同金属中心对反应中间体的选择性差异。
值得注意的是,所有催化剂的自由能路径中均未出现明显的能垒,表明反应在热力学上是可行的。此外,NO₃⁻的吸附强度与催化活性之间存在关联,Ti/g-C₃N₄的吸附最强但活性略低于V/g-C₃N₄,而Nb/g-C₃N₄的吸附和活性介于两者之间。
图7的数据还显示,NH₃的脱附自由能分别为0.71 eV(Ti/g-C₃N₄)、0.54 eV(V/g-C₃N₄)和0.18 eV(Nb/g-C₃N₄),表明Nb/g-C₃N₄更易于释放NH₃产物。
总体而言,通过自由能路径分析,明确了Ti/g-C₃N₄、V/g-C₃N₄和Nb/g-C₃N₄在NO₃RR中的反应机制和活性差异,其中V/g-C₃N₄因最低的极限电位(-0.25 V)和适中的吸附强度成为最具潜力的催化剂。
这些结果与全文的筛选结论一致,即单原子催化剂的金属中心特性(如d电子构型和电荷转移能力)显著影响NO₃RR的活性和选择性。