

电催化氮还原反应(NRR)作为可持续合成氨的关键技术,近年来在开发高效催化剂领域备受关注。在众多催化材料中,过渡金属合金凭借独特的电子结构和协同效应成为研究热点。
NiFe合金因其良好的电子结构调控能力和稳定性,在氮还原反应(NRR)催化中展现出广阔应用前景。通过调控Ni与Fe的比例,可优化d带中心位置,增强对N₂分子的吸附与活化能力,同时抑制竞争性的氢析出反应(HER)。合金界面形成的协同效应有助于电子在催化位点间的高效转移,提升中间体的反应动力学。
此外,NiFe合金具备良好的结构稳定性与导电性,适合在复杂电化学环境中长期运行。结合理论计算与实验研究,NiFe合金被认为是实现高效、可持续电催化氮还原的重要候选材料。


晶体结构与电子特性对NRR的影响
NiFe合金的催化性能与其晶体结构密切相关。根据Bradley相图(1937年),Ni-Fe合金的相结构随成分变化呈现显著差异:当Ni含量低于8%时形成体心立方(BCC)α相,而Ni含量超过32%时则形成面心立方(FCC)γ相。
例如,NiFe-500样品(500°C热处理)同时包含FCC和BCC结构,而NiFe-700样品(700°C)仅保留FCC结构,且后者在OER中表现出更低的过电位(307 mV)。
DFT计算表明,FCC结构的NiFe合金由于更紧凑的原子排列,其d带中心位置更接近费米能级,增强了氮气分子(N₂)的吸附和活化能力。
此外,Ni与Fe之间的电子转移(Ni的d-空位为0.6,Fe为2.2)进一步优化了活性位点的电子结构,促进N≡N键的断裂。
关键实验与理论验证
电子密度分布分析:在Ru1/mono-NiFe体系中,Ru原子的引入显著改变了NiFe的电子云分布,导致其态密度(DOS)峰值从-1.10 eV(块体NiFe)移动至-0.38 eV,表明更强的电子离域性,有利于N₂的吸附和电荷转移。
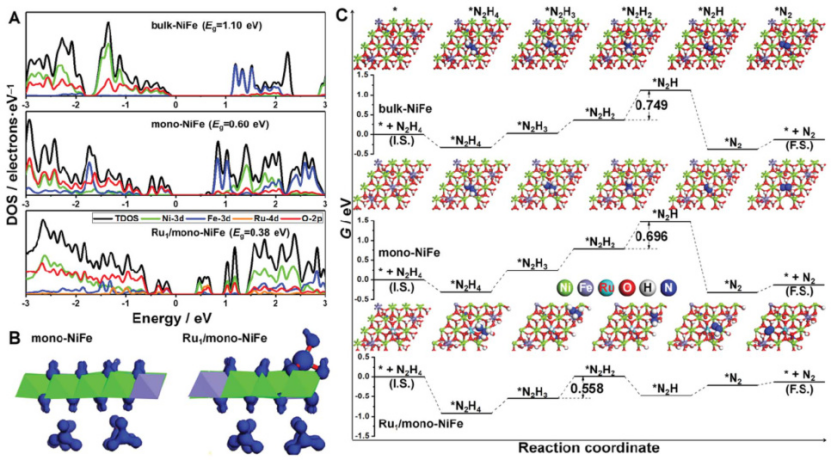
DOI: 10.1039/c8sc04480e
晶格畸变效应:NiFe与高熵合金(如FeNiMnCrCu)结合时,由于原子半径差异引起的晶格畸变增加了活性位点的暴露,并通过调节价电子浓度优化了N₂的吸附自由能。
NRR理论计算的核心方法与模型构建
DFT计算在NRR机理研究中占据核心地位,其流程通常包括模型构建、吸附能计算、反应路径模拟和自由能评估。以NiFe合金为例,关键步骤如下:
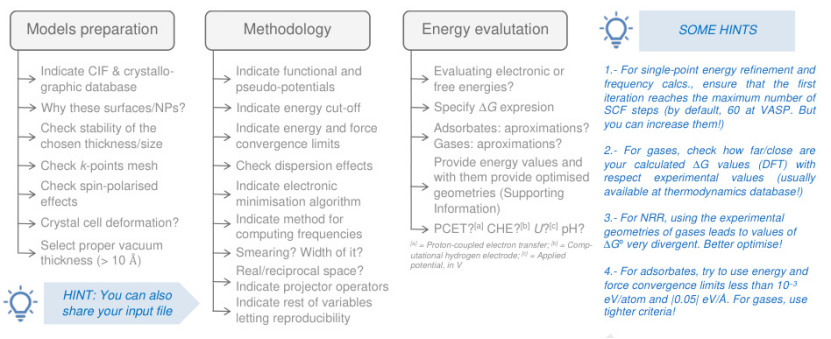
https://doi.org/10.1016/j.coelec.2022.101073
1. 模型准备:
在模型准备阶段,需合理选择催化剂的表面结构、几何参数及计算设置,以确保结果的准确性与物理意义。优先选取具有高指数晶面的NiFe(012)作为研究对象,可显著提高表面活性位点的密度。
为避免周期性边界条件对吸附行为产生干扰,真空层厚度一般设置在10 Å以上;此外,由于NiFe合金中Fe原子具有明显的自旋极化特性,需在计算中考虑自旋极化效应,尤其是在研究对磁矩敏感的N₂吸附构型时,其对能量及吸附态的影响不可忽略。
2. 吸附能与反应路径模拟:
在NiFe合金表面的N₂吸附过程中,常见的吸附模式包括端位(End-on)和侧位(Side-on)两种构型。端位吸附指N₂分子以一端与金属表面相连,而侧位吸附则是N₂分子以平行于金属表面的方式与多个金属原子相互作用。
研究表明,侧位吸附构型由于能与表面多个金属活性位点形成较强的电子相互作用,更有利于削弱N≡N三键的强度,从而促进其活化与后续的氮还原反应,具有更高的催化潜力。
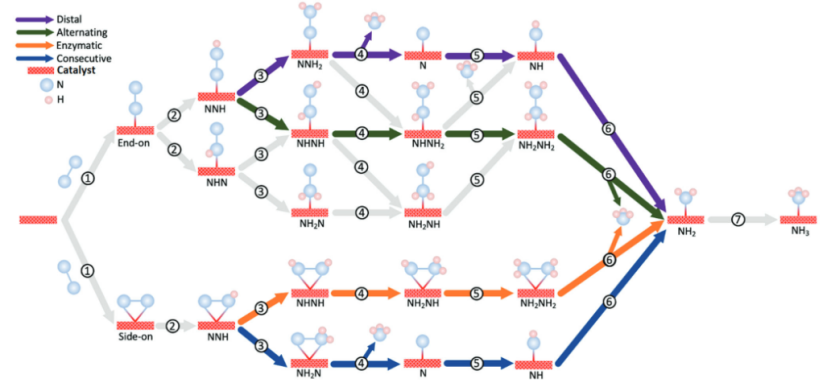
https://doi.org/10.1002/adma.202307150
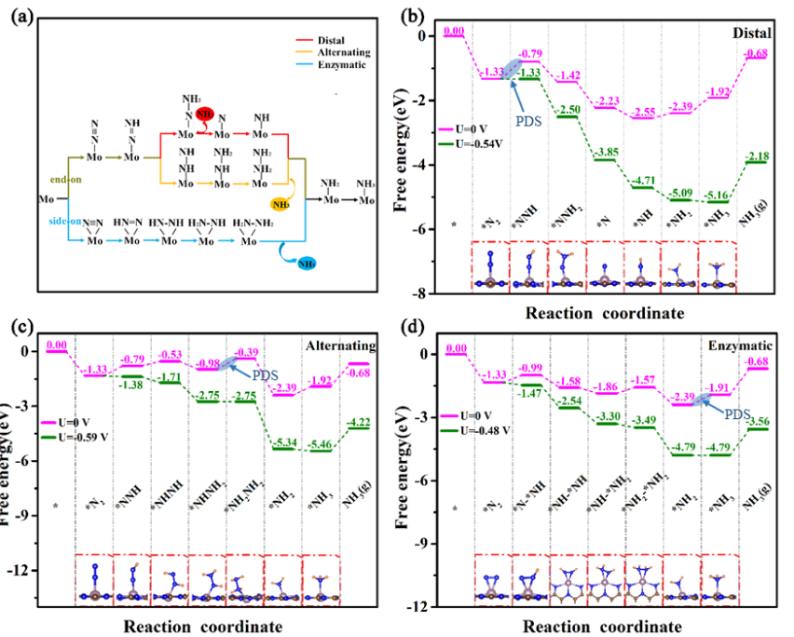
通过构建远端(Distal)、交替(Alternating)和酶促(Enzymatic)三种典型氮还原反应路径的自由能变化图,可以清晰地对比各反应路径的能垒差异。
在NiFe合金表面,计算结果表明交替路径中的关键限速步骤——N₂分子转化为NNH中间体(N₂ → NNH)具有最低的自由能障碍,这意味着反应更容易进行,有利于提高氮还原反应的催化效率。该结果为优化NiFe催化剂设计提供了重要理论依据。
https://doi.org/10.3390/molecules29194768
典型计算结果解析
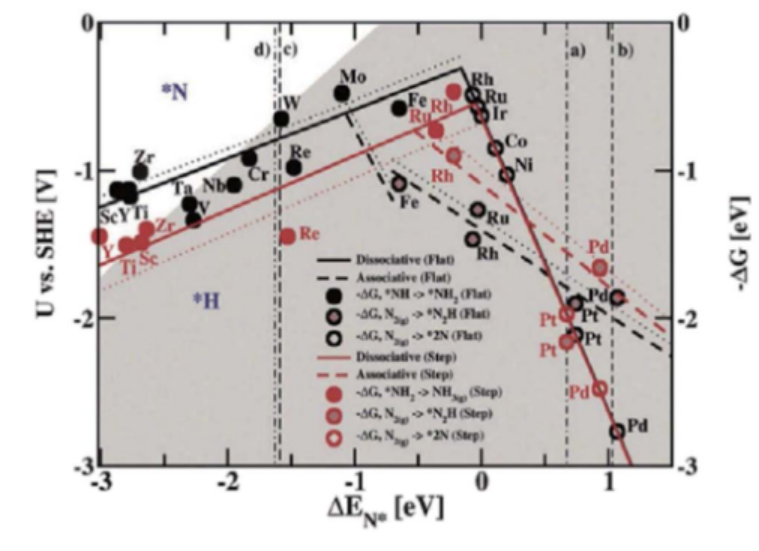
通过火山曲线分析可揭示催化剂氮吸附能与其催化活性之间的关系。
在以Fe为基准的氮还原反应火山图中,NiFe合金位于“解离型”反应路径的左侧区域,说明其对N原子的吸附强度适中(ΔE_N约为-0.5 eV),既能有效活化N₂分子,又避免中间体过度稳定而阻碍反应进程。
同时,NiFe合金表现出较低的理论过电位(约为-0.3 V vs. RHE),表明其在电催化氮还原反应中具有良好的活性与动力学优势,具备实际应用潜力。
DOI: 10.1039/d0ra08223f
NiFe基催化剂的NRR活性位点与反应机理
NiFe合金的活性位点识别是理论研究的重点。研究表明,Fe位点通常作为N₂吸附的初始活性中心,而Ni位点则促进后续的氢化步骤。
例如,在NiFe@V₂O₃复合结构中,V₂O₃基底通过强电子相互作用调控NiFe的电子态,使其表面形成富电子区域,从而增强N₂的吸附和活化。
https://doi.org/10.1021/acsomega.3c09920
关键文献案例解析
1. NiFe硒化物催化剂(Ni₀.₇₅Fe₀.₂₅Se₂):
通过层状双氢氧化物(LDH)衍生法合成的NiFe硒化物具有独特的纳米多孔结构,比表面积相比传统块体材料提高约三倍,有效提升了催化剂的表面活性位点密度。
DFT计算显示,N₂在NiFe-Se位点的吸附能为-0.45 eV,交替路径中限速步骤(NH₂ → NH₃)的自由能垒仅为0.78 eV,显著低于纯NiSe₂(1.2 eV)和FeSe₂(1.5 eV),表明其更优的氮还原反应动力学。
实际电催化测试中,在-0.1 V vs. RHE条件下,该材料实现了5.64 μg h⁻¹ cm⁻²的NH₃产率和12.3%的法拉第效率,整体性能优于多数贵金属催化剂,展现出良好的应用前景。
2. 双金属单原子催化剂(FeMo/g-C₃N₄):
Fe-Mo双金属位点通过形成供体–受体对实现协同催化机制,其中Fe的d轨道向N₂分子的反键轨道提供电子,增强其活化程度,而Mo的d轨道空穴则有助于质子耦合电子转移(PCET),共同降低反应的极限电位至-0.23 V,显著提升催化性能。
电荷密度差分析进一步表明,Fe-Mo位点的电荷转移量达ΔQ = 0.3 e⁻,远高于单一金属活性位点,清晰验证了双金属之间的协同效应在氮还原反应中的关键作用。
计算模型对比与新兴方法
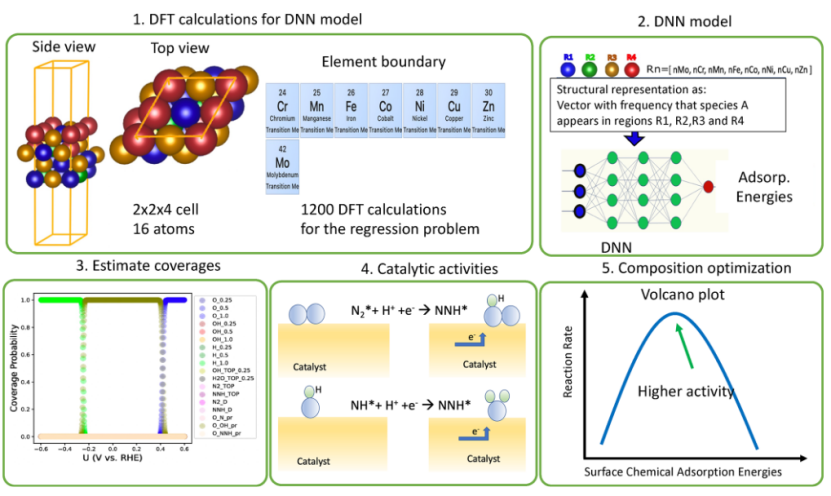
传统DFT计算虽能揭示反应机理,但面临计算成本高和动态过程模拟不足的挑战。近年来,神经网络与DFT的结合为NiFe合金设计提供了新思路:
1. 高熵合金(HEA)优化:
通过DFT计算构建FeNiMnCrCu等高熵合金(HEA)的吸附能数据库,结合神经网络模型进行组合筛选,有效预测出最优的合金成分。
基于此方法,Ru-TiNS合金经过神经网络优化后,其电催化合成氨的产率显著提升,达到15.19 μmol mg⁻¹ h⁻¹,是纯TiNS的约十倍,展示了机器学习辅助催化剂设计在提升性能方面的巨大潜力。
https://doi.org/10.1021/acscatal.3c05017
2. 动态过程模拟:
通过恒电位DFT模拟研究发现,在NiFe合金表面,N₂分子与质子(H⁺)的共吸附存在竞争关系,其中N₂优先吸附于低配位的Fe活性位点,有效抑制了氢气析出反应(HER)的发生,从而提升了氮还原反应的选择性。
此外,借助爬山弹道法(CI-NEB)对NiFe(012)晶面进行过渡态搜索,结果显示该晶面上O–O键耦合反应的能垒为1.51 eV,较常见的(110)晶面降低了0.6 eV,表明(012)晶面在催化过程中具有更优的动力学性能。
挑战与未来方向
尽管基于第一性原理的理论计算在NiFe合金氮还原反应(NRR)研究中已取得重要进展,但仍存在诸多挑战亟待解决。首先,NiFe催化剂表面在反应过程中可能发生结构重构,如转变为羟基氧化物NiFeOOH,因此需结合原位技术(如Operando XAFS)对理论模型进行实验验证。
其次,当前主要依赖原子级DFT计算,尚缺乏与介观尺度动力学模型的有效衔接,限制了对传质和相变等实际反应环境的准确描述。此外,借助图神经网络(GNN)等机器学习方法,可在大数据驱动下预测NiFe合金的吸附能与催化活性之间的关系,从而加速高效催化剂的筛选与优化。
https://doi.org/10.1021/acscatal.3c05017
总结
NiFe合金的NRR催化性能通过理论计算得以深入解析,其活性位点、电子结构调控和反应路径优化是核心研究方向。
结合DFT、火山曲线分析和机器学习,未来有望实现高效NiFe基催化剂的设计与合成,推动电化学合成氨技术的实际应用。