成键轨道(如σ、π)由原子轨道同相叠加形成,降低能量增强键合;反键轨道(如σ*、π*)异相叠加导致能量升高,削弱键强。
DFT通过COHP、COOP量化轨道贡献,如Fe₂Mo合金中Fe-Mo成键轨道优化氧还原反应(ORR)催化活性,降低能垒。
混合泛函(HSE06)提升反键轨道精度,机器学习加速COHP分析。未来结合动态轨道力(DOF)与多尺度模拟,推动催化与材料设计的精准调控。
成键轨道与反键轨道的定义
成键轨道(Bonding Orbital)和反键轨道(Anti-bonding Orbital)是分子轨道理论(Molecular Orbital Theory, MOT)中的核心概念:
成键轨道:由原子轨道的波函数同相叠加形成(线性组合为ψbonding=φ1+φ2),电子密度在两核之间显著增加,导致核间吸引力增强,能量低于原始原子轨道中能量最低者(例如,氢分子中σ成键轨道能量低于单个氢原子的1s轨道)。
反键轨道:由原子轨道波函数异相叠加形成(线性组合为ψanti-bonding=φ1−φ2),电子密度在核间区域减少,导致核间排斥力增强,能量高于原始原子轨道中能量最高者(例如,氢分子中σ*反键轨道能量高于单个氢原子的1s轨道)。
能量与稳定性的关键差异:分子轨道理论中,成键与反键轨道的能量差及其对称性特征是决定化学键稳定性的核心因素:成键轨道(如σ、π)通过原子轨道同相位叠加使能量显著降低(如H₂的σ轨道能量较原子轨道下降约8 eV),电子占据此类轨道时通过电荷密度在核间区域积累增强键合作用;
反键轨道(σ、π)则因轨道异相位叠加能量升高(如O₂的π轨道能量较原子轨道上升5 eV),其电子占据会削弱键强甚至导致解离(如O₂的顺磁性源于两个电子占据π轨道)。
轨道对称性方面,σ键具有圆柱对称性(如C₂H₄的C-C σ键),其成键轨道电荷密度沿键轴均匀分布;π键则呈现垂直于键轴的节面特征(如苯环的p轨道侧向重叠),其反键轨道π*在节面两侧形成相位相反的波瓣。
这种对称性差异直接影响分子反应活性,例如乙烯的π轨道电子易受亲电试剂攻击,而σ键因较高的空间屏蔽效应相对稳定。
成键/反键轨道组合的能级分布(如CO的5σ成键与2π*反键轨道)不仅决定分子稳定性,还调控其光电性质(如金属配合物的电荷转移跃迁能量),为分子设计与催化机理研究提供电子结构层面的理论依据。

DOI:10.1016/j.jpowsour.2025.236330
DFT计算中成键/反键轨道的分析方法
在密度泛函理论(DFT)框架下,成键与反键轨道的分析依赖于以下方法:
轨道能量与电子占据分析
密度泛函理论(DFT)通过求解Kohn-Sham方程获得分子轨道能量分布,其中成键轨道(如σ、π)通常占据较低能级(HOMO附近),反键轨道(σ、π)则位于较高能级(LUMO附近),例如CO的5σ成键轨道能量较C、O原子轨道下降6 eV,而2π*反键轨道上升4 eV。
态密度(DOS)与投影态密度(PDOS)分析可定量解析轨道贡献:在FeTe超导体中,Fe的e_g轨道(dz²、dx²−y²)成键态集中于-3~-1 eV能量区间,反键态分布于1~3 eV,其占据数(~1.2 electrons/orbital)通过Stoner判据触发铁磁不稳定性,而Te的p轨道在费米能级附近形成杂化态,调控超导能隙。
这种电子占据分析揭示材料宏观性质的内在机制,例如反键轨道部分占据(如O₂的π*轨道占据2电子)导致键级降低(O-O键级从2降至1),而全占据成键轨道(如N₂的σ与π轨道)赋予三键高稳定性。
通过轨道能量与占据数的关联,可指导催化剂设计(如调节d带中心优化吸附能)或光电材料开发(如调控带隙匹配太阳光谱)。
DOI:10.1016/j.apsusc.2022.153237
成键–反键相互作用量化工具
成键与反键相互作用的量化分析依赖COHP(晶体轨道哈密顿布居)、COOP(晶体轨道重叠布居)及动态轨道力(DOF)等工具:COHP通过哈密顿矩阵元分解量化原子间轨道相互作用的键合特性,正值表示成键贡献(如Fe-Fe金属键在-2~0 eV区间COHP>0),负值对应反键作用(如O₂的π*轨道COHP),其积分值(ICOHP)可定量比较键强度(如C-C单键ICOHP≈-3 eV,双键≈-5 eV);
COOP基于轨道重叠积分,在分子体系(如CO)中显示σ成键轨道(5σ)与π*反键轨道(2π*)的布居分布差异,指导前线轨道调控;DOF通过计算轨道能量对核位移的导数(∂ε/∂R),动态判断键合趋势,例如H₂解离过程中,当∂ε(σ)/∂R>0时轨道呈反键特性,触发键断裂。
这些工具在材料设计中广泛应用,如通过COHP分析FeTe超导体的Fe-Te键合强度(ICOHP=-2.1 eV)预测结构稳定性,或利用DOF优化催化剂活性位点的吸附构型(如Pt(111)表面CO吸附的轨道力分析),为揭示化学键本质与设计功能材料提供多维度量化指标。
DOI:10.1016/j.jmmm.2024.172502
软件与计算流程
成键与反键相互作用的定量分析需借助专业软件工具:Lobster作为后处理程序,可将VASP等DFT软件的平面波基组数据转换为原子轨道基组(如Fe的3d、4s和Te的5p轨道),通过投影重构晶体轨道哈密顿布居(COHP)与重叠布居(COOP)曲线。
例如,在FeTe超导体的COHP分析中,Fe-3d与Te-5p轨道在-3~0 eV能量区间呈现显著成键特征(COHP>0),而0~2 eV范围显示反键相互作用(COHP),其积分值(ICOHP=-2.1 eV)定量表征Fe-Te键强度。
对于复杂体系的轨道局域化分析,Wannier90软件通过最大局域化方法将Bloch波函数转换为Wannier轨道,如FeTe中e_g轨道(dz²和dx²-y²)经局域化后展现清晰的空间分布,其轨道能量分裂(Δ=1.5 eV)揭示晶体场效应导致的能级劈裂。
这种局域轨道可视化不仅可识别层状材料中的面内σ键与层间π键(如石墨烯的sp²杂化轨道),还能解析拓扑绝缘体的表面态轨道特征(如Bi₂Se₃的Dirac锥形成机制)。
结合Lobster与Wannier90的计算流程(结构优化→电子自洽→轨道投影→局域化分析),研究者能系统揭示材料的成键本质与电子输运特性,为设计新型超导、拓扑材料提供原子级理论指导。
实例分析: Fe₂Mo合金
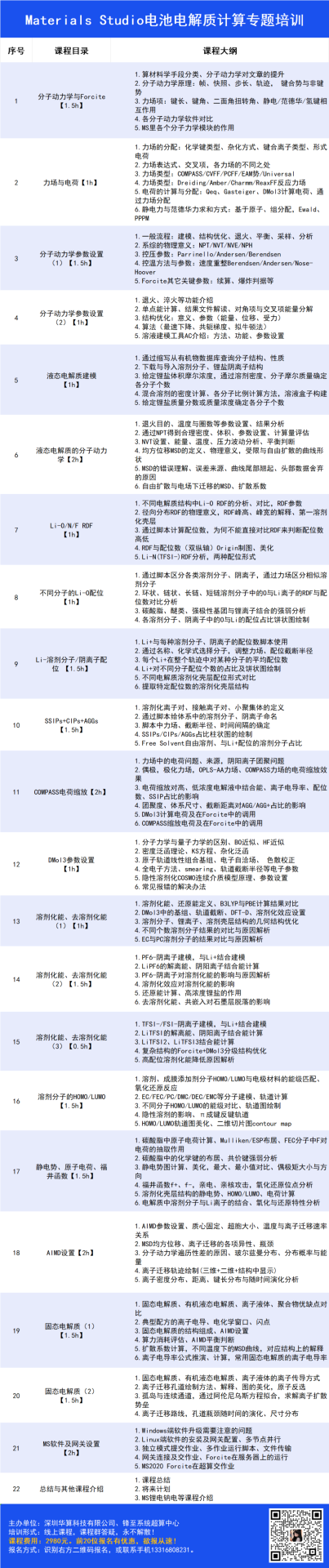
在《FeMo双金属合金促进酸碱环境下高效稳定 ORR 催化》的研究中,作者借助密度泛函理论(DFT)深入剖析了 Fe₂Mo 合金的成键与反键轨道特性及其对氧还原反应(ORR)催化性能的影响机制。
通过轨道分布分析发现,费米能级附近的成键轨道(蓝色)主要集中于 Fe 与 Mo 原子之间,形成强烈的轨道耦合区域,而反键轨道(绿色)则分布于原子外围(如图 4a 所示),这种空间分布特征表明 Fe-Mo 原子间存在显著的轨道重叠与电子离域效应,进而通过强相互作用稳定了合金的电子结构,为催化反应提供了稳定的活性位点基础。
进一步的电子态分析通过 Fe-3d 和 Mo-4d 轨道的投影态密度揭示,低能区的成键轨道电子占据数显著高于高能区的反键轨道,表明 Fe 与 Mo 的 d 轨道在成键过程中贡献了更多的共享电子,形成稳固的金属 – 金属键合。
这种电子结构特征不仅增强了 Fe-Mo 原子间的协同作用,还通过调整活性位点的电子自旋密度与电荷分布,有效降低了 ORR 关键步骤的反应能垒。
在 ORR 反应路径的能量分析中,Fe₂Mo 合金在酸性和碱性环境下均展现出较低的过电势,其核心机制归因于成键轨道对反应中间体(如 * O、OH、OOH)吸附能的优化。
成键轨道的强电子耦合使 Fe 位点的 d 带中心位置调整,既避免了中间体过度吸附导致的位点中毒,又防止了吸附过弱引发的反应动力学迟滞,从而在双电层环境中实现了中间体吸附 – 脱附过程的平衡。
例如,在碱性条件下,Mo 原子通过反键轨道的电子反馈效应调节 Fe 位点的氧化态,削弱O 的吸附强度,使决速步能垒显著降低;而在酸性环境中,Fe-Mo 的轨道杂化增强了对OOH 中间体的稳定作用,促进了氧 – 氧键的断裂。
这种酸碱环境下的普适性优势,本质上源于成键轨道主导的电子结构调控与反键轨道辅助的吸附能优化的协同效应。
该研究通过轨道分布、电子态密度与反应路径能量的多维度 DFT 分析,揭示了双金属合金中原子间轨道耦合对催化活性的关键影响:成键轨道的强相互作用不仅稳定了活性位点结构,还通过电子离域效应优化了中间体吸附行为,而反键轨道的空间分布特征则在调控电子自旋与电荷转移中发挥辅助作用。
这些发现为理解双金属催化剂的协同机制提供了原子尺度的理论依据,也为设计兼具高效性与环境适应性的 ORR 催化剂指明了方向 —— 通过合理调控金属间的成键 / 反键轨道分布,可实现活性位点电子结构与吸附性能的精准优化,从而突破单一金属催化剂在复杂电化学环境中的性能局限。
DOI:10.1007/s12274-022-4112-1
总结
在成键与反键轨道的密度泛函理论(DFT)分析中,高精度泛函开发与计算方法创新显著提升了对复杂电子结构的描述能力。
混合泛函(如 HSE06)通过结合局域和非局域交换关联能,有效改善了对反键轨道(如 π* 轨道)能量位置及形状的描述精度,而长程修正方法(如 DFT-D3)则通过引入色散校正,优化了弱相互作用体系中轨道相互作用的计算准确性。
在此基础上,机器学习技术的融入为成键性质分析带来新突破:利用神经网络模型可高效预测晶体轨道哈密顿布居(COHP)和晶体轨道重叠布居(COOP)曲线,快速解析复杂体系中原子间的成键强度与轨道重叠特征,极大加速了传统 DFT 计算中耗时的成键分析过程。
动态轨道力(DOF)方法进一步扩展了 DFT 在激发态动力学领域的应用,通过结合非绝热耦合效应,该方法能够捕捉激发态演化过程中轨道的成键 – 反键转变行为,为光诱导化学反应中键的断裂与形成机制提供动态视角。
这些方法通过态密度分析、COHP/COOP 曲线量化及动态轨道力模拟,构成了从静态电子结构到动态键合过程的完整理论框架,成为理解化学键本质、催化反应路径及材料电子性质的核心工具,为催化活性位点设计、功能材料能带调控等提供了精准的理论指导。
随着计算硬件与算法的持续进步,未来成键 / 反键轨道分析将通过高精度泛函、机器学习与多尺度模拟的深度融合,实现对复杂体系键合行为的更精确预测与高效设计,推动理论化学与材料科学研究从定性描述向定量调控的范式转变。
写在最后
热门电解质计算方法在MS锂电池电解质课程中均有讲解。
课程基于Materials Studio Forcite,DMol3, Amorphous Cell,CASTEP模块设计,既包含液态、固态电解质的设计,也涵盖溶剂分子的电子性质研究。