

说明:本文华算科技系统介绍了如何利用DFT计算研究MXene材料的结构、稳定性、电子特性及表面吸附行为,从结构优化、稳定性预测到能带分析和吸附能计算的全套方法,帮助实验高效设计高性能MXene材料,加速能源与催化领域的研究与应用。
什么是MXene材料?
MXene是一类新型的二维过渡金属碳化物、氮化物和碳氮化物,其通式为Mn+1XnTx(其中M为过渡金属,X为碳或氮,Tx为表面终止基团)。
MXene因其独特的物理化学性质(如高电导率、可调控的电子结构、优异的机械性能和丰富的表面化学)而在能源存储、催化、传感和电子器件等领域展现出巨大应用潜力。
随着计算材料科学的飞速发展,理论计算与模拟已成为研究MXene材料不可或缺的工具,不仅能够深入理解其内在性质,还能指导实验设计和新材料开发。
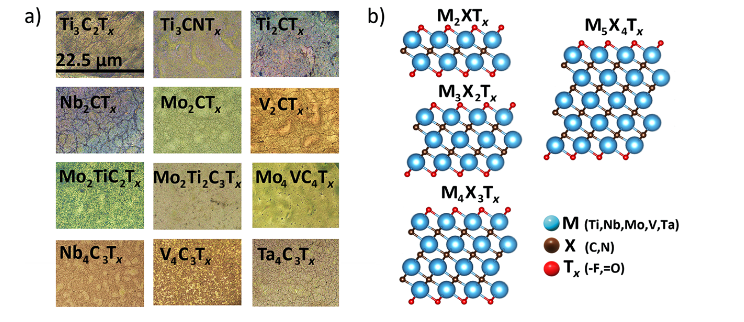

DOI:10.1021/acs.chemmater.4c01536
结构优化与稳定性预测计算
MXene的结构优化是其所有计算研究的基础步骤,旨在找到能量最低的稳定几何构型。优化过程通常采用共轭梯度法或BFGS算法进行离子弛豫,同时保持晶胞参数可变以适应MXene层间的可能变化。
在DFT计算中,收敛标准的设置至关重要:能量收敛阈值通常设置为10-5-10-6 eV/原子,而力收敛阈值则设置为0.01-0.02 eV/Å,确保原子受力足够小以达到稳定构型。
平面波截断能是影响计算精度和效率的关键参数,对于含有过渡金属的MXene体系,通常设置为400-600 eV,以平衡计算精度和成本。
k点网格采样则采用Monkhorst-Pack方法,对于典型的MXene单层结构,通常使用15×15×1到24×24×1的网格密度确保布里渊区积分的准确性。
由于MXene是二维材料,必须在垂直方向添加足够的真空层(通常大于15Å)以避免周期性镜像层间的非物理相互作用。
MXene的稳定性评估涉及多个方面:热力学稳定性、动力学稳定性和热稳定性。形成能计算是评估热力学稳定性的基本手段,通过比较MXene与其组成元素(或前驱体MAX相)的能量差来判断其合成可行性。
负的形成能表明该结构在热力学上有利,但还需进一步验证其动力学稳定性。
DOI: 10.1039/D1MA00474C
声子谱计算是评估动力学稳定性的金标准,通过Phonopy等工具计算整个布里渊区的声子色散关系,确保无虚频(负频率)存在。
例如,对硼基MXene衍生物的计算研究表明,某些构型虽然在热力学上稳定,但声子谱中存在虚频,表明其动力学不稳定性,可能无法在实验中合成。
从头算分子动力学(AIMD)模拟用于研究MXene的热稳定性,通常在300-500K温度下进行几个ps的模拟,观察其结构是否发生明显变形或分解。
例如,对Ti₃C₂Tₓ MXene的AIMD模拟表明,即使在500K高温下,其基本骨架仍能保持稳定,但表面终止基团可能发生旋转或交换,这解释了实验中观察到的MXene热稳定性差异。
近年来,机器学习势函数与高通量计算相结合的方法大大加速了MXene稳定性预测。通过USPEX等结构预测算法与机器学习结合,能够高效筛选潜在稳定的MXene组成,如对硼基MAX相和MXene衍生物的预测研究所示。
电子结构性质计算
MXene的电子结构计算主要关注能带结构和态密度(DOS),这两者是理解其电学、光学和磁学性质的基础。计算能带结构时,需要沿布里渊区的高对称路径进行能量本征值计算,通常采用更密的k点网格(如36×36×1)以确保精度。
对于带隙计算,标准GGA-PBE泛函往往低估带隙值,因此需采用HSE06混合泛函或GW方法进行修正,以获得与实验相符的结果。
态密度(DOS)计算包括总态密度和投影态密度(PDOS),后者能够分解出不同原子轨道(s、p、d、f)对电子态的贡献。通过分析PDOS,可以识别MXene中金属-碳/氮键的杂化情况,以及表面终止基团对费米能级附近电子态的影响。
例如,对铜基MXene的计算研究表明,其总态密度在费米能级附近有显著分布,表明金属性行为,而部分态密度(pDOS)显示Cu-d轨道在费米能级处起主导作用。
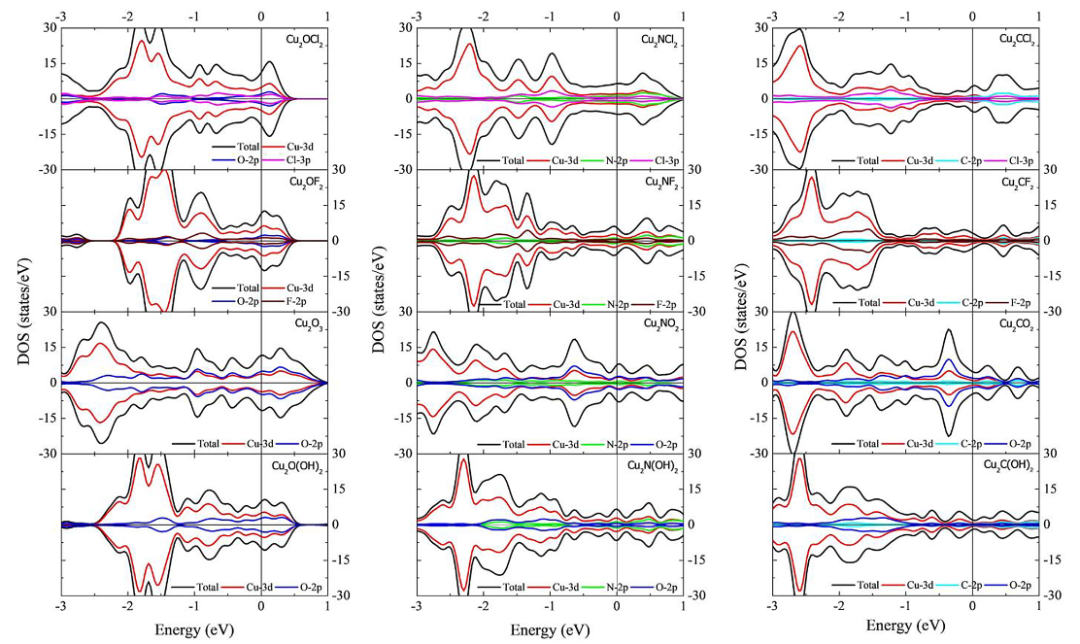
DOI:10.1038/s41598-021-90628-2
表面吸附计算
MXene的高比表面积和丰富表面化学使其成为优异的吸附和催化材料,其表面吸附行为主要通过吸附能(Eₐd)来量化。吸附能计算公式为:Eₐd = EMXene+adsorbate – EMXene – Eadsorbate,其中负值表示放热吸附过程,且越负表示吸附越强。
DFT计算能够精确预测不同吸附质(如H₂O、CO₂、NH₃、H₂等)在MXene表面的吸附构型和吸附能,为理解吸附机制提供原子级洞察。
吸附位点是影响吸附行为的关键因素,MXene表面通常存在三种可能位点:顶位(top site,位于表面原子正上方)、桥位(bridge site,位于两个表面原子之间)和空心位(hollow site,位于多个原子形成的中心)。
不同吸附质倾向于占据不同位点,例如水分子通常通过氧原子与表面金属原子形成配位键,优先占据顶位;而氢分子则可能在空心位发生解离吸附。
除了吸附能和吸附位点,其他关键参数还包括:电荷转移(通过Bader电荷或Mulliken布居分析量化)、功函数变化、态密度移以及吸附前后结构变化等。这些参数共同揭示了吸附过程的物理化学本质,为设计高性能吸附材料提供指导。
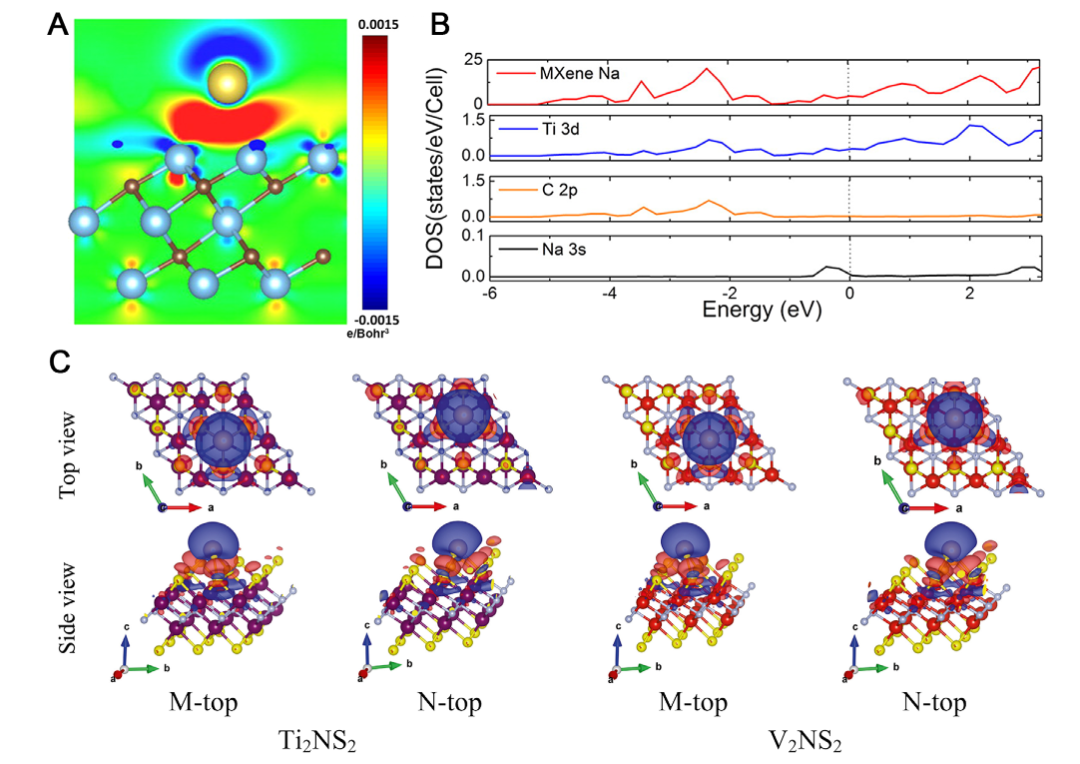
DOI:10.1002/EXP.20210024
结论
随着计算方法的不断进步和计算资源的日益丰富,理论计算将在MXene研究中发挥越来越重要的作用,不仅能够深入理解其基础物理化学性质,还能预测新功能、设计新材料、优化器件性能,最终实现从”计算指导实验”到”计算即实验”的范式转变。
【做计算 找华算】
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服
务。?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???