

说明:本文华算科技旨在系统阐述界面能的物理定义及其重要性,并重点探讨如何利用基于密度泛函理论(DFT)的维也纳从头算模拟软件包(VASP)进行精确的理论计算。
界面能的物理定义与科学意义
界面能,有时也称为界面自由能或界面张力,是一个关键的热力学概念。其最核心的定义是:在恒温恒压下,可逆地创建一个单位面积的界面所需要做的非体积功。
换言之,它是指与构成界面的两个体相材料相比,界面区域所具有的单位面积的过剩能量。这种能量过剩源于界面处的原子与体相内部的原子相比,其化学环境、配位数和成键状态发生了改变,导致体系能量升高。从数学上,界面能(γ) 可以表示为:
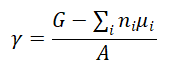

其中,G是含有界面的整个体系的总吉布斯自由能,ni和μi分别是组分的摩尔数和化学势,A是界面的面积。在理论计算中,常在0K下进行,此时吉布斯自由能近似等于总能量。需要注意的是,表面可以看作是界面能的一个特例,即材料与真空形成的界面所对应的能量。
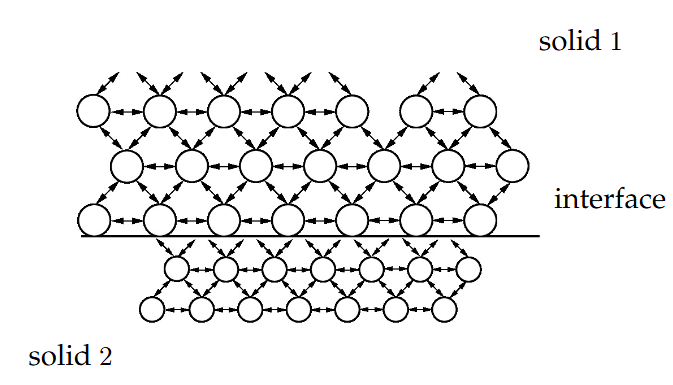
基于VASP的界面能第一性原理计算
VASP作为一款强大的第一性原理计算软件,通过求解Kohn-Sham方程来获得材料的电子结构和总能量,为精确计算界面能提供了理论工具 。其计算核心在于精确获得包含界面的超胞体系与相应体材料的能量差。
核心计算公式
对于一个由A和B两种材料构成的界面,其界面能 (γAB) 的计算通常采用“Slab模型”(或称板模型)。在一个沿着某个方向(如z轴)具有周期性边界条件的超胞中,我们构建一个包含A/B界面的结构。由于周期性,该超胞通常包含两个对称的A/B界面。因此,界面能的计算公式为
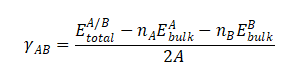
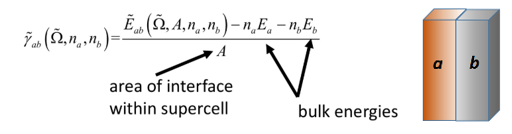
其中:
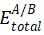 是包含A/B界面的超胞经过充分结构优化后的总能量
是包含A/B界面的超胞经过充分结构优化后的总能量
![]() 和
和![]() 分别是构成界面的材料A和B的体材原胞的总能量
分别是构成界面的材料A和B的体材原胞的总能量
nA和nB分别是超胞中材料A和B的化学式单数量。
A是界面的横截面积。
分母中的“2”是因为该模型通常包含两个等价的界面。
标准计算工作流程
步骤一:构建超胞模
这是计算成功与否的关键第一步。首先,需要对材料A和B的体结构进行优化,获得平衡晶格常数。然后,选择合适的晶面来构建界面。为了减小由于晶格失配引入的应变,通常需要对A和B的晶格进行扩胞,使其在界面平面内(如x-y平面)的晶格矢量尽可能匹配,应变一般控制在2%以内。
随后,将匹配好的A和B的Slab结构堆叠起来,并在z方向上加入足够厚度(通常 > 15 Å)的真空层,以消除周期性边界条件带来的镜像相互作用。Slab本身也需要有足够的厚度,以保证其中部原子呈现出体材料的性质。
DOI:10.1063/5.0238437
步骤二:计算各组分能量
体材料能量计算:对优化好的材料A和B的体材料原胞进行高精度的静态自洽计算,得到能量 和
和![]()
界面体系能量计算:对构建好的A/B界面超胞模型进行结构优化。通常会固定超胞中远离界面的几层原子,只允许界面附近区域的原子进行弛豫,以模拟体材料对界面的“钳制”效应。优化完成后,进行一次高精度的静态自洽计算,得到总能量。
步骤三:计算界面能
将上述计算得到的能量值代入核心计算公式,即可求得界面能。
结论
界面能是连接材料微观结构与宏观性能的核心物理量。基于VASP等第一性原理计算软件,研究人员能够通过构建精细的原子模型和精确的总能量计算,从量子力学层面揭示界面能的物理本质。
一个严谨的界面能计算工作流,不仅要求对计算原理有深刻理解,更需要在模型构建、参数选择和收敛性测试等环节进行细致和审慎的操作。通过理论计算与实验现象的紧密结合,我们可以更深入地理解界面行为,从而为高性能新材料的设计与开发提供有力的理论指导。