

本文聚焦密度泛函理论(DFT)在异质结构界面能研究中的应用。
首先阐释界面能的物理内涵与DFT计算公式,揭示其与化学键合、电荷重排的关联;进而通过案例说明DFT在预测界面稳定性(如G/BN堆叠构型)、优化力学性能(金属/陶瓷复合材料)、调控电子结构(肖特基二极管)及设计催化活性位点(CO₂还原)中的作用。
研究表明,DFT为异质结构的微观机制解析与宏观性能优化提供了关键理论支撑,助力材料在电子、催化等领域的创新应用。
引言
在材料科学与工程领域,异质结构作为一种由两种或两种以上不同材料在原子尺度上结合而成的复合体系,正逐渐成为推动技术革新的核心力量。
从纳米电子器件中精准调控载流子输运的半导体异质结,到高效分解水制氢的光催化异质结构,界面工程通过对异质结构中材料间界面性质的精确设计与优化,实现了单一材料难以企及的优异性能。
以半导体器件为例,硅/锗异质结的引入显著提升了晶体管的开关速度和电子迁移率,推动了集成电路向更小尺寸、更高性能方向发展;在催化领域,金属 – 氧化物异质结构通过界面协同效应,极大增强了催化剂对目标反应的活性和选择性,为能源转化与环境治理提供了新的解决方案。
然而,异质结构界面的微观物理化学过程极为复杂,实验手段在直接测量和深入理解界面能等关键参数时面临诸多挑战,这为第一性原理计算,特别是密度泛函理论(Density Functional Theory,DFT)的应用提供了广阔空间。
DFT 基于量子力学原理,从电子结构层面出发,通过求解薛定谔方程的近似形式,能够在原子尺度上精确计算材料的各种性质,而无需依赖经验参数。在异质结构界面能研究中,DFT 的独特优势得以充分展现。
实验方法如表面张力测量、界面断裂能测试等,往往只能获得宏观尺度的平均结果,难以捕捉原子级界面的精细结构与电子态变化;此外,部分极端条件下(如高温、高压)的界面性质实验测量难度极大甚至无法实现。
相比之下,DFT 能够在计算机中构建理想或缺陷界面模型,系统探究不同原子排列、化学成分和外部条件对界面能的影响,为理解界面形成机制、预测材料性能提供理论依据,从而有效弥补实验研究的局限性,成为连接材料微观结构与宏观性能的桥梁。
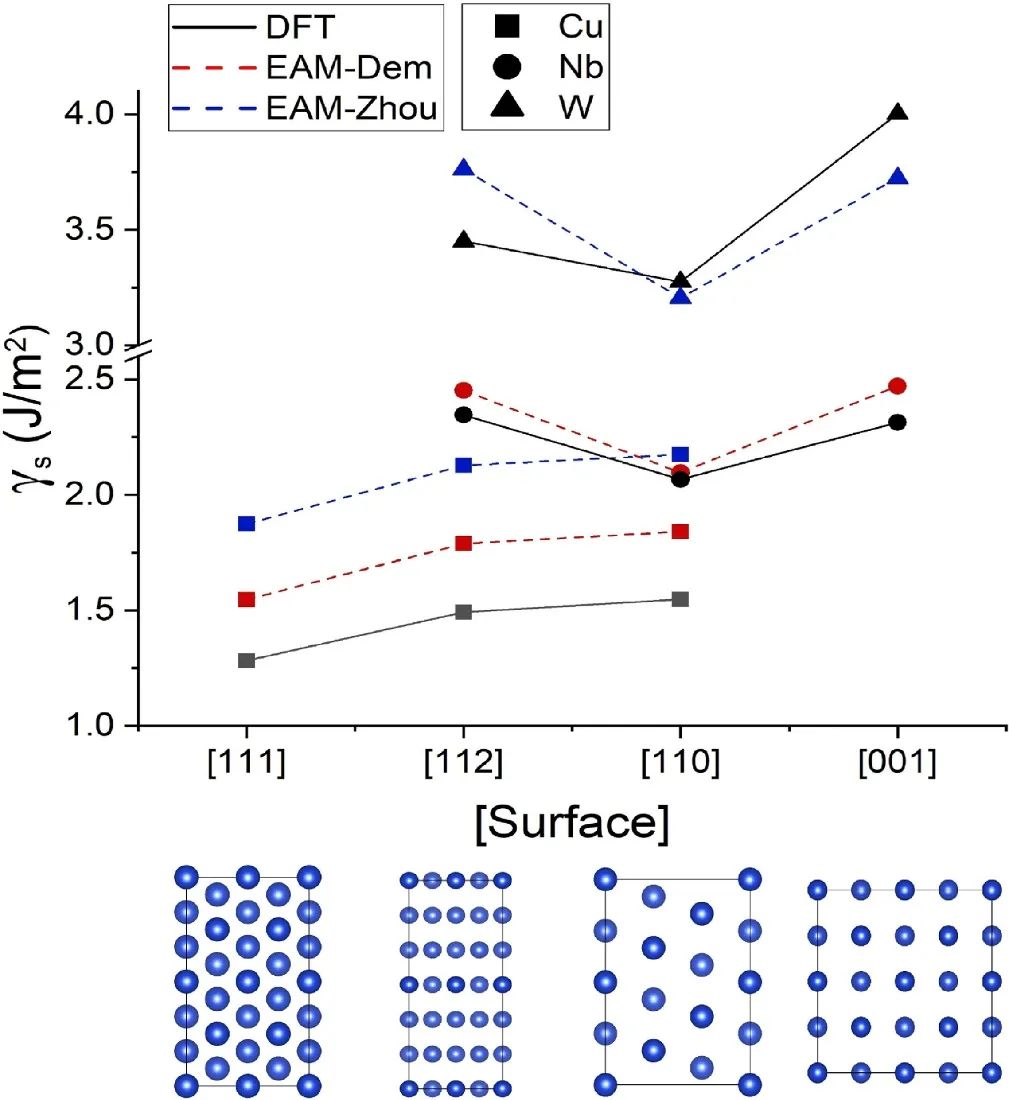

界面能的DFT定义与物理内涵
什么是界面能
界面能是描述异质结构中材料界面性质的关键物理量,本质上反映了体系在形成界面过程中能量的变化。从微观角度来看,当两种不同材料结合形成异质界面时,原子间的化学键发生重构,电子云重新分布,体系的总能量也随之改变。
界面能的大小不仅决定了异质结构的热力学稳定性,还深刻影响着材料的力学、电学、光学等性能。较低的界面能意味着界面形成过程释放能量,体系更倾向于自发结合;反之,较高的界面能则表明界面处于亚稳态,存在结构重组或分离的趋势。因此,准确理解和计算界面能,是掌握异质结构性质、实现材料性能优化的重要前提。
基本公式
在 DFT 计算中,异质结构界面能的计算公式为:
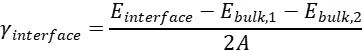
其中,为异质结构总能量,和为两材料的体相能量,为界面面积。
关键物理意义
界面能作为异质结构稳定性的核心判据,其数值正负直接反映了界面形成过程的热力学趋势。当界面能 0 ,则意味着界面形成需要外界提供能量,体系处于不稳定状态,界面存在分离趋势。
从化学键合角度来看,界面能与原子间的相互作用密切相关。在共价键主导的异质界面(如碳纳米管 – 石墨烯异质结)中,界面能的大小取决于原子轨道的重叠程度和电子云的共享程度;离子键界面(如金属氧化物 – 卤化物异质结构)的界面能则主要受离子电荷分布和静电相互作用的影响;而在金属键界面(如金属合金异质结构)中,自由电子气的重新分布对界面能起决定性作用。
此外,界面处的电荷重排也是影响界面能的重要因素。当两种材料形成异质界面时,由于电子亲和能和功函数的差异,电子会在界面两侧重新分布,形成电荷转移,这种电荷重排不仅改变了界面的电子结构,还通过库仑相互作用影响界面能大小。例如,在半导体异质结中,电荷转移形成的内建电场会显著改变界面能,进而影响载流子的输运特性。
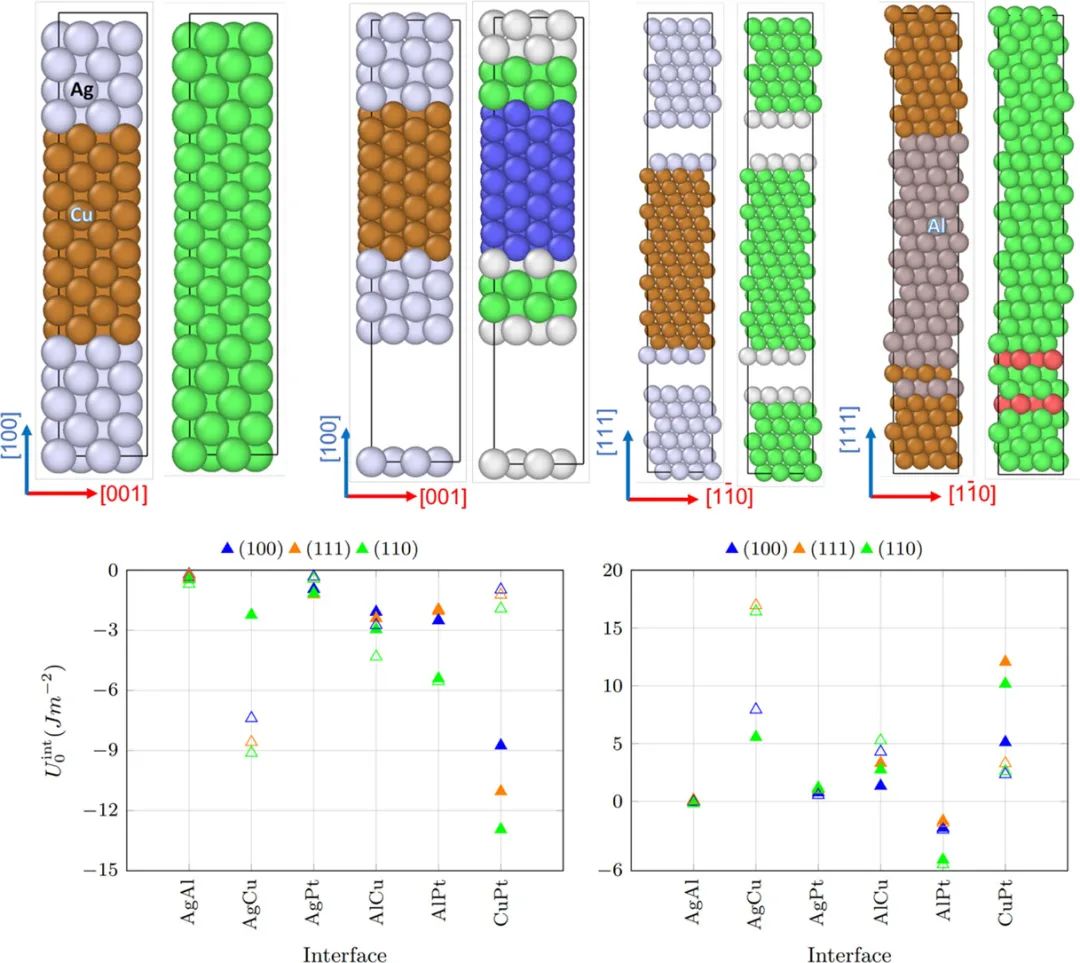
界面能的应用场景
界面稳定性预测
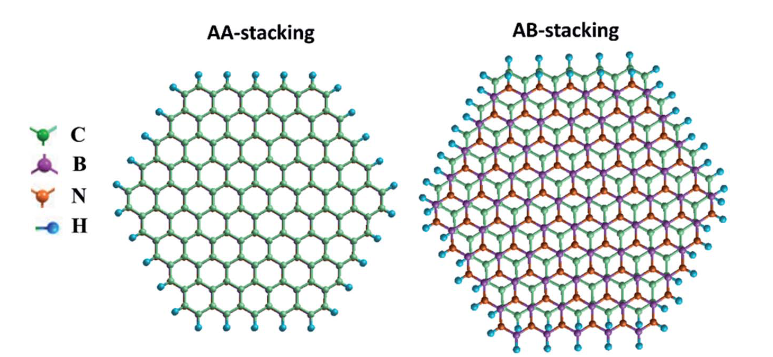
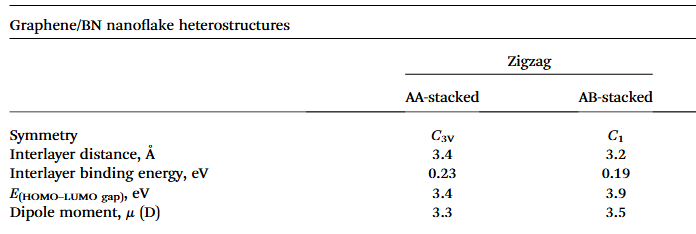
在异质结构设计中,预测不同堆叠构型的界面稳定性是关键环节。以石墨烯 / 氮化硼(G/BN)异质结构为例,其存在 AA 和 AB 等多种堆叠方式,不同堆叠构型下的界面能差异显著影响着异质结构的稳定性和物理性质。
DFT 计算表明,AA 堆叠的 G/BN 异质结构界面能相对较高,体系处于亚稳态,而 AB 堆叠构型具有较低的界面能,热力学稳定性更强。这是由于 AB 堆叠时,石墨烯和氮化硼的原子层间形成更紧密的范德华相互作用,电子云分布更为匹配,从而降低了体系能量。
通过 DFT 系统计算不同堆叠角度和层间距下的界面能,能够为实验制备提供精确的理论指导,帮助研究者选择最稳定的堆叠构型,避免因构型选择不当导致的材料性能不稳定问题。此外,DFT 还可用于研究缺陷(如空位、掺杂)对界面稳定性的影响,为设计具有特定功能的异质结构提供理论依据。
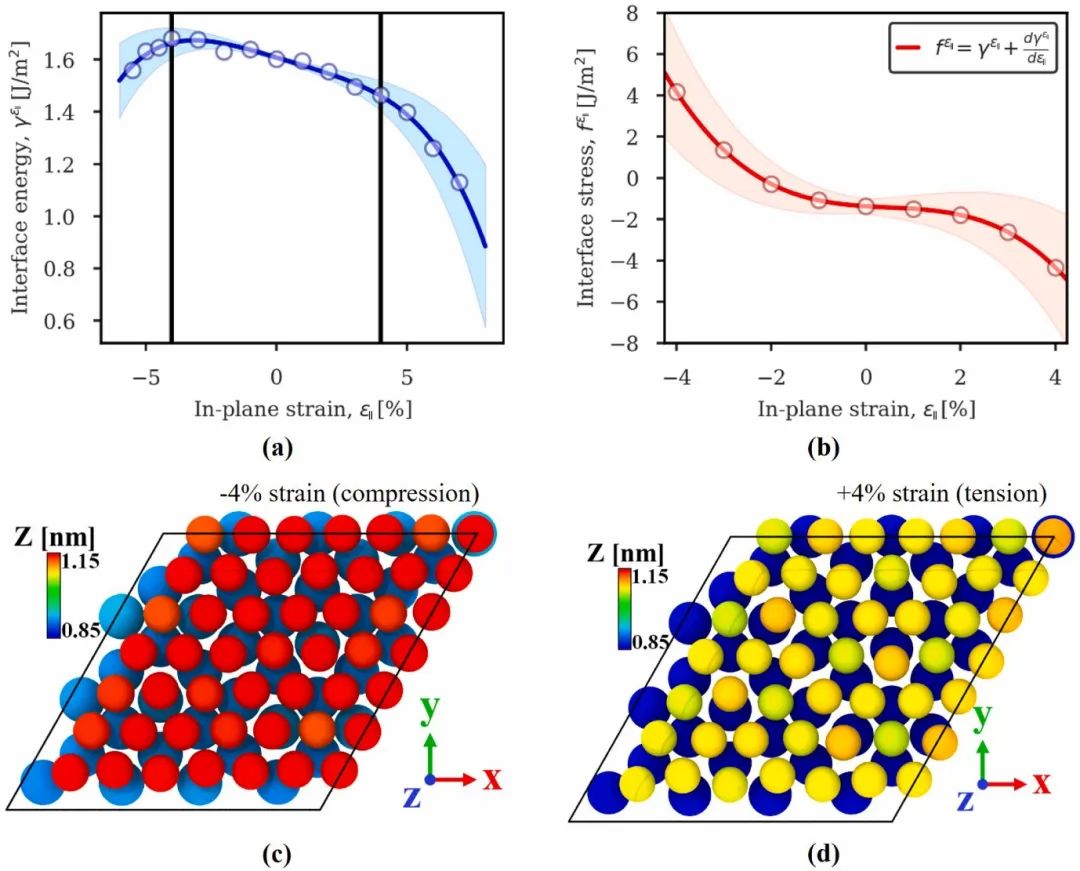
力学性能优化
界面能与异质结构的力学性能,尤其是断裂韧性密切相关。在金属 / 陶瓷复合材料中,金属相和陶瓷相的界面能决定了裂纹在界面处的扩展行为。较低的界面能意味着界面结合较弱,裂纹更容易沿着界面扩展,导致材料整体韧性下降;而较高的界面能则使界面结合增强,裂纹扩展时需要消耗更多能量,从而提高材料的断裂韧性。
通过 DFT 计算不同界面结构(如原子排列、界面相组成)下的界面能,并结合分子动力学模拟裂纹扩展过程,可以深入理解界面能对力学性能的影响机制。
例如,在钛合金 / 氧化铝陶瓷复合材料中,通过 DFT 设计引入过渡金属层,调节界面原子间的化学键合强度,降低界面能的同时增强界面结合力,有效抑制裂纹扩展,显著提升了复合材料的断裂韧性和强度。这种基于 DFT 的设计策略为高性能复合材料的开发提供了理论指导,推动了其在航空航天、汽车制造等领域的广泛应用。
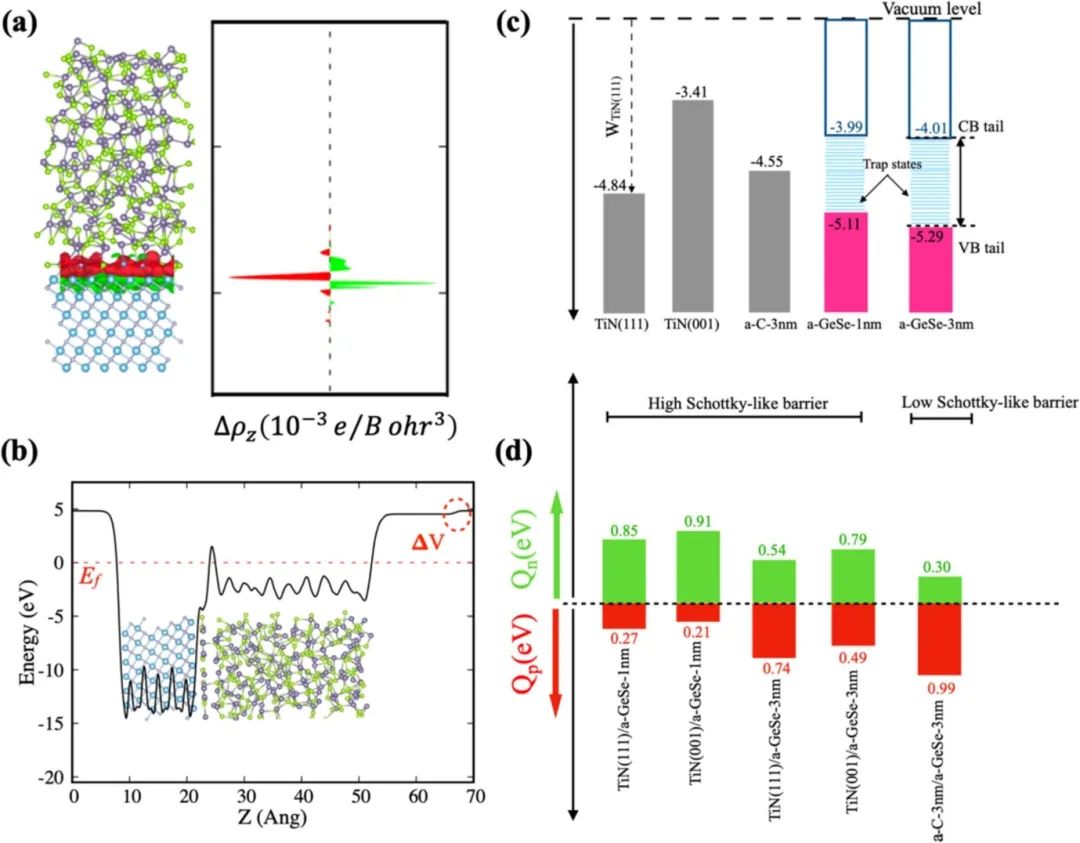
电子结构调控
在半导体器件设计中,异质结构界面的电子结构调控至关重要,而界面能与电荷转移密切相关,直接影响着器件的电学性能。以肖特基势垒二极管为例,金属与半导体形成的异质界面处,电子从费米能级较高的材料向费米能级较低的材料转移,形成空间电荷区和肖特基势垒。
DFT 计算能够精确分析界面处的电荷分布、能带对齐和肖特基势垒高度,揭示界面能与电子结构之间的内在联系。通过改变金属材料种类、半导体掺杂浓度或引入界面修饰层,利用 DFT 计算不同条件下的界面能和电子结构变化,可以实现对肖特基势垒的精准调控。
例如,在硅基肖特基二极管中,通过 DFT 计算发现,在金属 / 硅界面引入超薄氧化物层,能够有效降低界面能,改变电荷转移程度,进而减小肖特基势垒高度,提高二极管的整流性能和开关速度。这种基于 DFT 的电子结构调控策略为高性能半导体器件的设计与优化提供了重要的理论支撑。
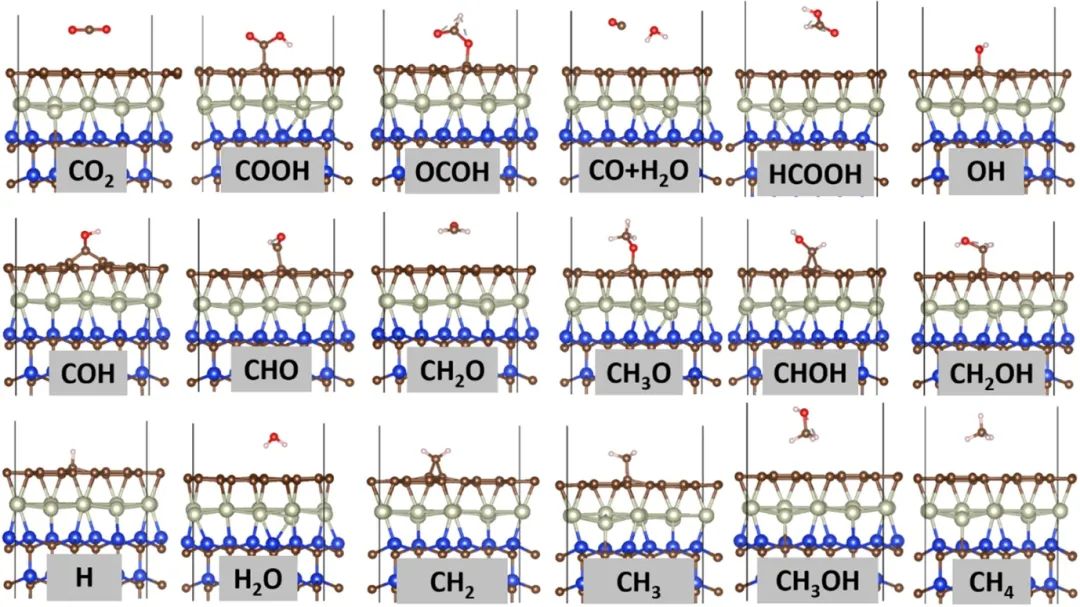
催化活性位点设计
在催化领域,异质结构界面能可作为活性位点暴露程度和催化活性的有效描述符。以二氧化碳(CO₂)还原反应为例,催化剂的活性位点通常位于界面或表面缺陷处,界面能的大小反映了活性位点的稳定性和可及性。
DFT 计算能够模拟不同界面结构下的 CO₂吸附、活化和转化过程,通过计算吸附能、反应能垒等参数,结合界面能分析,筛选出具有高催化活性的异质结构。例如,在金属 – 氧化物异质结构催化剂中,DFT 研究表明,较低的界面能有利于活性金属原子的暴露和稳定,促进 CO₂分子在界面处的吸附和活化,降低反应能垒,从而提高催化活性。
此外,通过 DFT 设计界面结构,引入特定的缺陷或掺杂原子,调节界面能和电子结构,可进一步优化活性位点的性质,增强催化剂对目标反应的选择性。这种基于 DFT 的催化活性位点设计策略,为开发高效、高选择性的催化剂提供了理论指导,推动了能源催化领域的发展。
综上所述,DFT 在异质结构界面能研究中发挥着不可替代的重要作用。通过精确计算界面能及其相关物理量,DFT 不仅帮助我们深入理解异质结构界面的微观物理化学过程,还为材料设计与性能优化提供了有力的理论工具。
随着计算方法的不断改进和计算机性能的提升,DFT 在异质结构研究领域将展现出更大的应用潜力,有望为解决能源、电子、催化等领域的关键科学问题提供新的思路和解决方案。
未来,结合实验表征和机器学习等技术,构建多尺度、多学科交叉的研究体系,将进一步拓展 DFT 在异质结构界面能研究中的应用范围,推动材料科学向更高水平发展。
上述文案从多维度展现了 DFT 在异质结构界面能研究中的应用。你对内容深度、案例选择等方面若有其他想法,欢迎随时提出。