

本文系统介绍了在DFT计算过程中确定晶面的两种最实用方法。第一种是基于表面能计算的方法,通过比较不同晶面的表面能来预测最稳定的晶面结构。第二种是结合实验表征技术(如XRD和HRTEM)来确定晶面,将实验观测结果作为DFT建模的基础。
文章详细说明了每种方法的适用场景、实施步骤和注意事项,为材料模拟研究中的晶面选择提供了明确的指导。这两种种方法可单独使用,也可相互验证,共同确保DFT计算中晶面选择的准确性。
计算表面能确定晶面
原理
对表面能计算结果的深入分析是理解晶体材料表面性质的关键。表面能作为衡量晶体表面稳定性的重要物理量,其数值大小直接反映了晶面的相对稳定性。
从物理本质上讲,低表面能的晶面意味着原子在该晶面上的排列更加紧密和有序,原子间的相互作用更强,电子云分布更加均匀和稳定。这种稳定的原子排列和电子结构使得晶面具有较低的能量状态,在热力学上更倾向于保持稳定,不易发生变化。
例如,对于金属晶体,(111)晶面通常具有较低的表面能,这是因为在(111)晶面上,原子呈密堆积排列,原子间的距离最小,相互作用力最强,从而导致该晶面的表面能较低。
相反,高表面能的晶面原子排列相对松散,原子间的距离较大,相互作用力较弱,电子云分布也不够稳定。这种不稳定的结构使得晶面具有较高的能量,在热力学上处于相对不稳定的状态。
高表面能晶面更容易与周围环境发生相互作用,例如吸附外来分子、发生化学反应等,以降低自身的能量,达到更稳定的状态。在晶体生长过程中,表面能的差异起着决定性作用。晶体总是倾向于沿着表面能较低的晶面生长,因为这样可以使整个晶体体系的总能量降低,从而达到更稳定的状态。
这就导致在晶体生长过程中,低表面能晶面会逐渐扩大,而高表面能晶面则会逐渐缩小甚至消失。因此,通过分析表面能计算结果,可以深入了解晶体的生长习性和表面结构的形成机制。
案例
这篇文献通过密度泛函理论(DFT)计算金属氧化物(Co₃O₄、α-Fe₂O₃和In₂O₃)不同晶面的表面能,结合Wulff构造模型和断键密度指数(D₆),系统研究了晶体形态的热力学调控机制及其在材料科学中的应用价值。
研究首先基于Wulff定理,通过表面能计算预测了晶体在平衡状态下的形貌特征,例如Co₃O₄的立方体、八面体及伪八面体形态,与实验观测结果高度吻合。表面能作为决定晶体生长方向的关键参数,其相对大小直接影响了各晶面的暴露比例,而DFT计算为这一热力学过程提供了定量依据。
Co₃O₄的(111)晶面因表面能最高(2.31 J/m²)且断键密度最大(53.17 nm⁻²),在催化反应中表现出最优活性,这与实验中(111)晶面主导的样品显示最高电催化性能的结论一致。
此外,通过分析α-Fe₂O₃的(001)-O晶面,研究发现表面氢钝化可显著降低其能量(0.782 J/m²),揭示了表面修饰对稳定性的调控作用,为实验中选择合成条件(如溶剂、pH值)提供了理论指导。
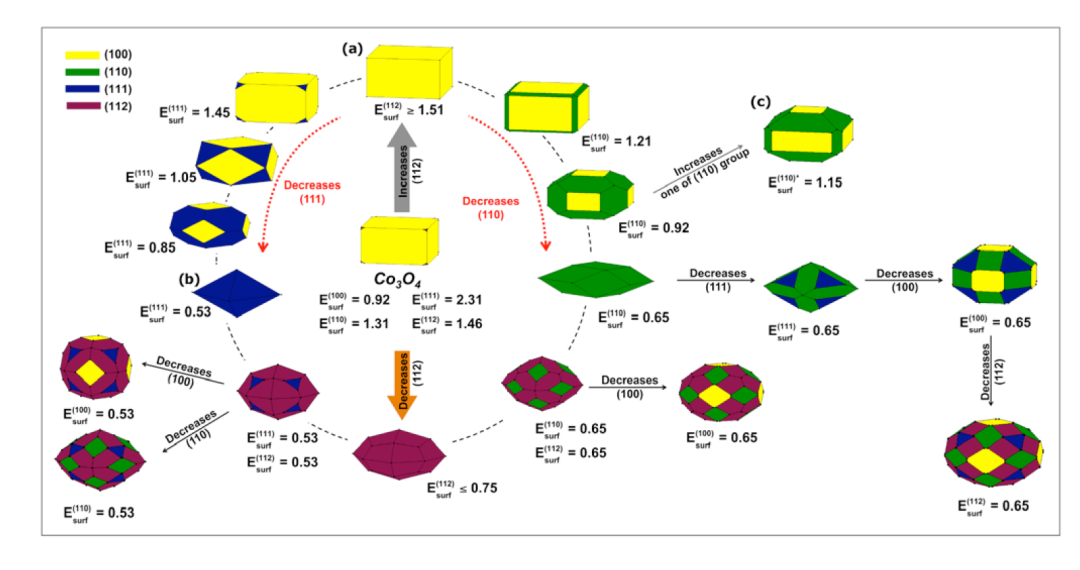

进一步地,研究阐明了晶体形态动态转变的原子机制。通过调整表面能的相对比值,如改变反应温度或添加表面活性剂,可实现从立方体到截角八面体的可控转变。
例如,α-Fe₂O₃在十二烷基胺(ODA)辅助合成中,随着ODA浓度增加,(112)和(001)晶面占比上升,形成截角十四面体,与Wulff构造模拟的形态演变路径完全匹配。这种理论预测能力不仅减少了实验试错成本,还深化了对“表面能驱动生长”这一核心原理的理解。
对于In₂O₃,计算表明(100)晶面能量最高(1.759 J/m²),导致其在生长过程中逐渐消减,最终晶体以(110)和(111)晶面为主,与实验室合成的多面体形貌一致,验证了理论模型的普适性。
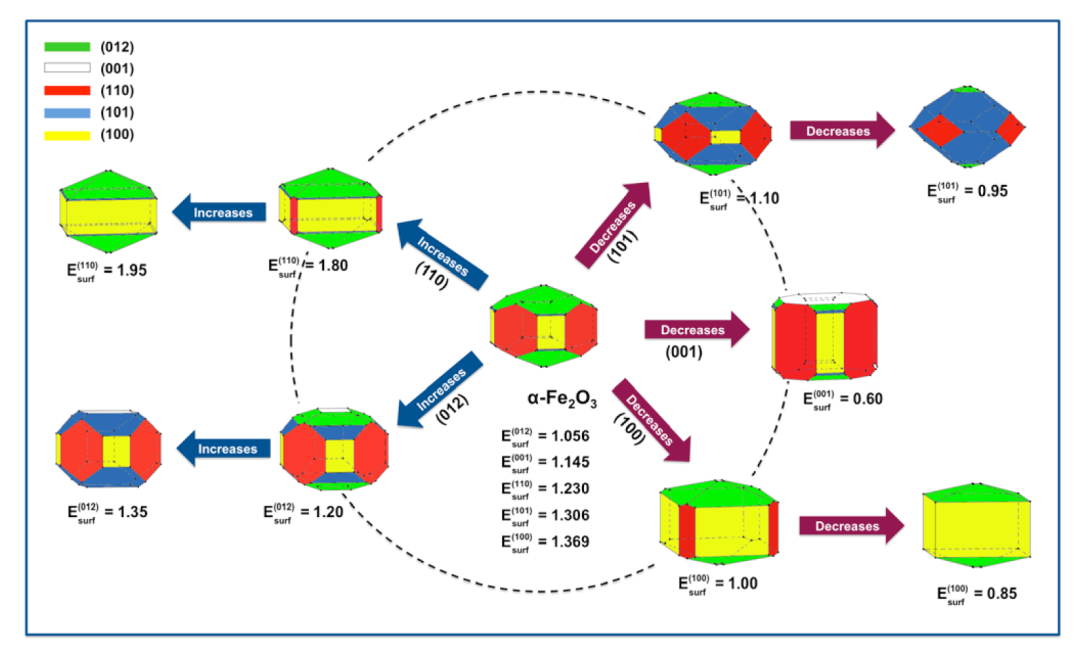
研究还强调了DFT计算在跨尺度关联中的桥梁作用。通过将原子级别的表面能数据与宏观形貌特征关联,解决了实验难以直接测量表面能的难题。
例如,Co₃O₄的(110)晶面在特定电位下因表面能变化(1.31 J/m²)而优先暴露,从而优化了锂–氧电池的电化学性能,这一发现为设计高效电极材料提供了新思路。
此外,断键密度指数(D₆)的引入建立了表面原子配位不饱和度与稳定性的直接联系,如α-Fe₂O₃的(001)-Fe晶面虽表面能较高(1.145 J/m²),但因断键密度最大(54.65 nm⁻²),在特定合成条件下仍可稳定存在,这解释了实验中观察到的复杂形貌多样性。

该研究的核心贡献在于将理论计算、热力学模型与实验验证紧密结合,不仅证实了表面能作为形态调控的主导因素,还提出了“通过表面化学调控实现形貌设计”的通用策略。
例如,通过模拟Co₃O₄不同晶面的表面能分布,可预判添加羧酸类表面活性剂会选择性抑制(100)晶面生长,从而获得高活性(111)晶面暴露的催化剂。这种理性设计方法显著提升了材料性能优化的效率和针对性。
文献最后指出,尽管当前计算未考虑溶剂化或氧化还原环境影响,但其建立的框架已为后续研究(如界面效应、动态环境模拟)奠定了基础。
总之,该工作通过DFT计算表面能,不仅揭示了金属氧化物形态调控的物理化学本质,还为功能材料的定向合成提供了理论工具。其意义不仅限于解释实验现象,更在于指导如何通过调控表面化学来设计具有特定形貌和性能的纳米材料,从而推动催化、能源存储、传感器等领域的应用发展。
未来,结合更复杂的环境效应计算和机器学习方法,这一研究范式有望进一步拓展至多元化合物和动态生长过程的精准预测。
化学键理论与分子轨道的诞生
原理
在密度泛函理论(DFT)计算中,准确选择晶面是模拟材料表面性质的关键。X射线衍射(XRD)和透射电子显微镜(TEM/HRTEM)作为一对相辅相成、互为补充的实验表征手段,在为晶面确定提供可靠依据方面发挥着极为重要的作用。
XRD基于布拉格方程,通过对材料衍射图谱的细致分析,能够精准地解析出材料的晶面间距以及复杂的晶体结构。凭借其快速高效的检测特性,它能够在较短时间内筛选出材料中主要暴露的晶面。由于XRD信号源自材料的整体衍射效应,这就使得其检测结果具备广泛的统计代表性,特别适用于初步确定材料的优势晶面。
在实际操作过程中,将实验测得的XRD图谱与标准衍射数据库进行详细比对,能够有效地排除杂质相的干扰,从而确保后续建模所采用的晶面与实验样品完全一致,为研究的准确性奠定坚实基础。
案例
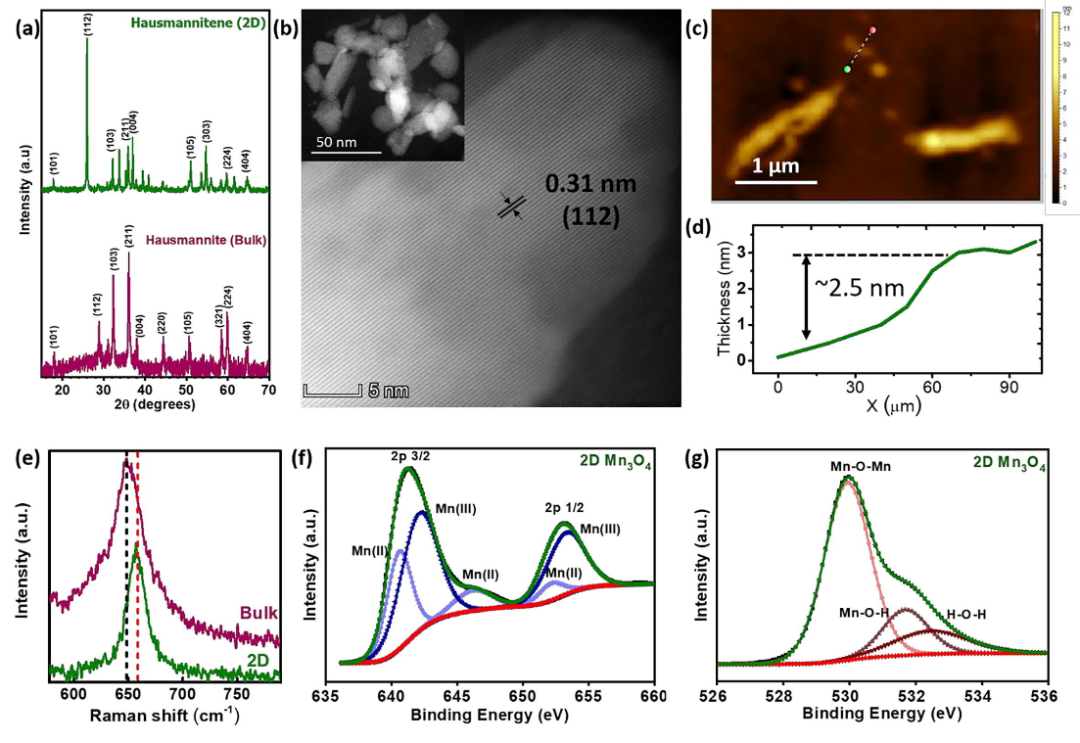
这篇文献通过系统的实验表征和理论计算相结合的方法,深入研究了超薄二维Mn₃O₄在电催化氧析出反应(OER)和氧还原反应(ORR)中的活性机制。
首先,作者通过高分辨透射电子显微镜(HRTEM)观察到Mn₃O₄的晶格条纹间距约为0.31nm,对应于Mn₃O₄ 的(112)晶面,这一结果与X射线衍射(XRD)数据中(112)晶面衍射峰的增强和位移现象相互印证,表明(112)晶面在二维结构中占据主导地位。
选区电子衍射(SAED)进一步验证了(112)晶面的单晶性质,为后续理论模型的构建提供了可靠的实验基础。在密度泛函理论(DFT)计算部分,作者首先优化了体相Mn₃O₄的晶格参数(a=5.89 Å, b=5.89 Å, c=9.57 Å),与文献报道值高度一致。
基于TEM和XRD的实验结果,作者选择了(112)晶面作为研究对象,构建了一个2×1×1的超胞模型,其晶格参数为a=8.09Å, b=14.0Å, c=25.70Å,以充分体现表面效应。
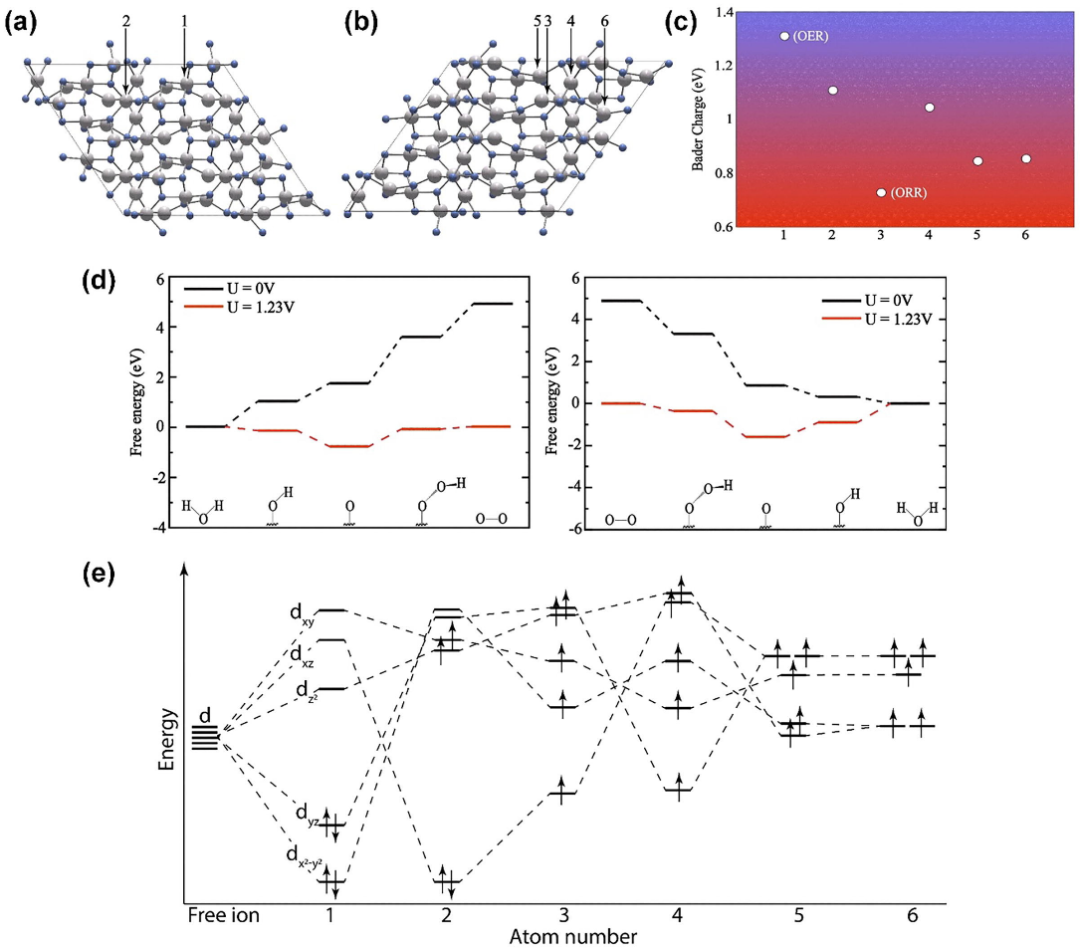
为了深入理解(112)晶面的催化活性起源,作者进行了Bader电荷分析,计算了表面Mn原子的电荷分布,发现Mn(1)和Mn(3)位点分别具有最低(0.72)和最高(1.31)的正电荷,因此被确定为ORR和OER的潜在活性位点。
通过定性晶体场理论(CFT)分析,作者进一步揭示了这些位点的电子结构特性:Mn(III)位点的最低未占据轨道(dz²)能量较低,易于接受电子,表现出优异的OER活性;而Mn(II)位点的最高占据轨道(dxz)能量较高,易于提供电子,展现出良好的ORR活性。
为了验证这些位点的催化性能,作者优化了OER和ORR反应中间体(*OOH、*O、*OH)在(112)表面的吸附构型,并计算了它们的吸附自由能(ΔG)。通过构建自由能图,作者确定了OER和ORR的速率决定步骤,并计算出理论过电位分别为0.85eV和0.9eV,表明(112)晶面具有较低的反应能垒和较高的催化效率。
实验表征与理论计算的紧密结合为理解Mn₃O₄的高催化活性提供了全面的视角。X射线光电子能谱(XPS)分析显示,Mn₃O₄中Mn(III)的含量(64.87%)显著高于体相材料(31%),这与DFT预测的Mn(III)位点作为OER活性中心的结果高度一致。
此外,TEM和XRD确认的(112)晶面主导性为DFT模型的构建提供了坚实的实验依据,而DFT计算则从电子结构层面解释了该晶面高催化活性的内在机制。
这种多尺度、多角度的研究方法不仅揭示了Mn₃O₄(112)晶面在OER和ORR中的关键作用,还为设计高效、稳定的二维氧化物电催化剂提供了新的思路和理论指导。通过实验与理论的协同验证,作者成功建立了材料结构–活性关系,为未来开发新型催化材料奠定了重要基础。