

在电催化研究中,d带中心(d-band center)是一个重要的电子结构参数,用于描述过渡金属表面的电子结构特性,并与催化活性密切相关。通过第一性原理计算(如VASP),可以准确计算d带中心,从而为催化剂设计和性能优化提供理论支持。以下将详细介绍如何使用VASP计算d带中心,包括计算步骤、关键参数设置、数据处理方法以及实际应用案例。



一、d带中心的基本概念
d带中心是指过渡金属原子的d轨道电子密度在费米能级附近的平均位置。它反映了金属原子的电子结构特性,特别是在吸附反应中的作用。d带中心的计算通常基于态密度(DOS)和投影态密度(PDOS)的分析。PDOS能够更精确地反映特定原子轨道的电子分布,因此在计算d带中心时,通常需要使用投影态密度数据。
二、计算d带中心的步骤
1. 结构优化与自洽计算
在计算d带中心之前,需要对催化剂表面进行结构优化,并进行自洽计算。这一步骤确保了计算结果的准确性。在VASP中,可以通过设置ISIF=3和IBRION=2进行结构优化,使用LREAL=Auto来加速计算。完成自洽计算后,会生成一个vasprun.xml文件,其中包含电子结构信息。
2. 投影态密度(PDOS)的计算
为了计算d带中心,需要从DOSCAR文件中提取投影态密度数据。DOSCAR文件包含了每个原子的投影态密度信息,可以通过pymatgen库进行处理。具体步骤如下:
安装pymatgen:如果尚未安装,可以通过pip安装:
pip install pymatgen
读取DOSCAR文件:使用pymatgen读取DOSCAR文件,并提取投影态密度数据:
from pymatgen.electronic_structure.core import Dos
dos = Dos.from_file(“DOSCAR”)
提取d轨道数据:假设目标原子是第0个原子(如Pd原子),可以提取其d轨道的投影态密度:
d_center = dos.get_d_band_center()
结果输出:d_center将返回d带中心的值,单位为eV。
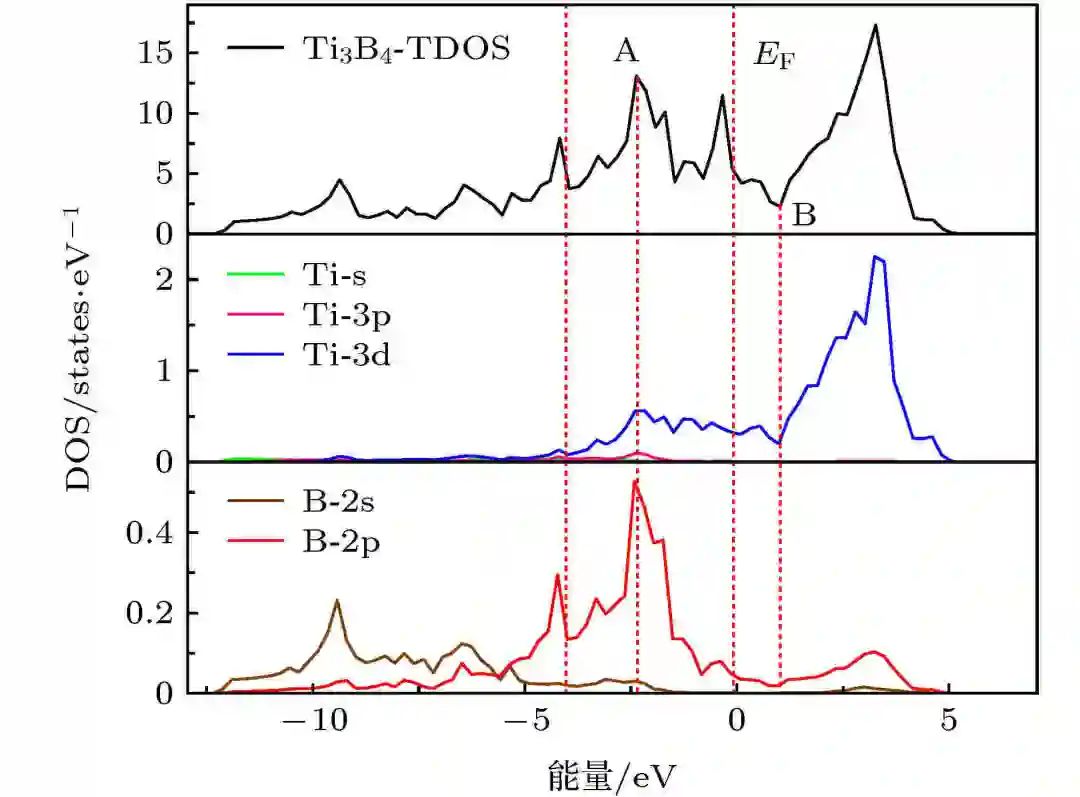
三、d带中心与催化活性的关系
d带中心与催化活性之间存在线性关系。研究表明,d带中心越接近费米能级,吸附能力越强,催化活性越高。例如,在Co4N和V-Co4N催化剂中,V掺杂后d带中心向更负的方向偏移,导致吸附能力减弱,但通过调整d带中心位置,可以有效提升催化活性。
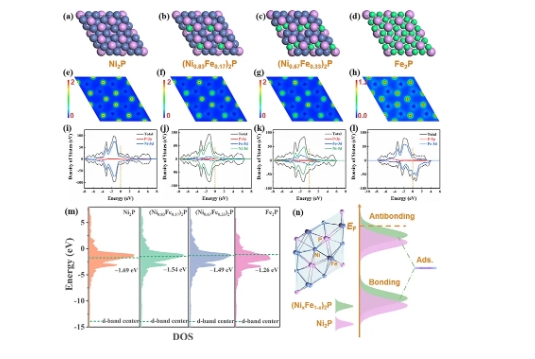
四、实际应用案例
1. Pd(111)表面的d带中心计算
在Pd(111)表面,d带中心的计算结果为-1.59 eV。这表明Pd原子的d轨道电子密度在费米能级附近,具有较强的吸附能力。通过计算不同表面的d带中心,可以评估其在HER、OER等反应中的催化性能。
2. Co4N催化剂的d带中心分析
在Co4N催化剂中,d带中心的计算结果为-1.79 eV。通过掺杂V元素,d带中心向更负的方向偏移,导致吸附能力减弱。然而,通过调整d带中心位置,可以有效提升Co4N的HER催化活性。
3. Cu(221)面的d带中心分析
在Cu(221)面,d带中心的计算结果为-1.0 eV。通过分析不同表面的d带中心,可以评估其在CO2RR反应中的催化性能。
五、总结
通过VASP计算d带中心,可以深入理解过渡金属表面的电子结构特性,并与催化活性建立联系。计算步骤包括结构优化、投影态密度提取、数据处理和可视化。实际应用中,d带中心与催化活性之间存在线性关系,通过调整d带中心位置,可以优化催化剂性能。未来的研究可以进一步探索d带中心与多电子效应、自旋效应等复杂现象的关系,为催化剂设计提供更全面的理论支持。