

说明:在蛋白质研究领域,准确预测活性位点对于解析蛋白质功能、药物设计等至关重要。
以下六大专业网站为该领域提供了有力工具:CASTp可定位并测量蛋白质表面口袋与内部空腔的几何参数,FTMap 通过碎片探针识别结合热点区域,HotSpot Wizard 整合多源数据注释蛋白质工程 “热点” 残基,P2Rank基于机器学习算法对配体结合位点进行高精度评分与聚类,ConSurf 通过进化保守性分析揭示功能关键区域,COACH作为元服务器整合多种算法与数据库实现结合位点的综合预测。
这些工具凭借各自的技术优势,覆盖了从结构几何分析到功能进化注释、从单一算法预测到多源数据整合的多维度需求,为科研人员探索蛋白质活性位点提供了多样化的高效解决方案。本文将给大家详细介绍这六个网站。
CASTp

地址:http://sts.bioe.uic.edu/castp/
CASTp (Computed Atlas of Surface Topography of proteins) 是一个功能强大的在线数据库和计算工具,专门用于识别、计算和可视化蛋白质三维结构(主要来自PDB数据库)表面的空腔、口袋和隧道。
它使用精确的数学方法(基于拓扑学的α-shape算法)来计算这些表面特征,提供它们精确的几何和拓扑性质,包括体积、表面积、开口大小、深度以及组成其表面的氨基酸残基信息。
该网站为研究人员理解蛋白质功能位点(如配体结合位点、酶活性位点、蛋白质相互作用界面)以及辅助基于结构的药物设计提供了关键的三维结构信息。
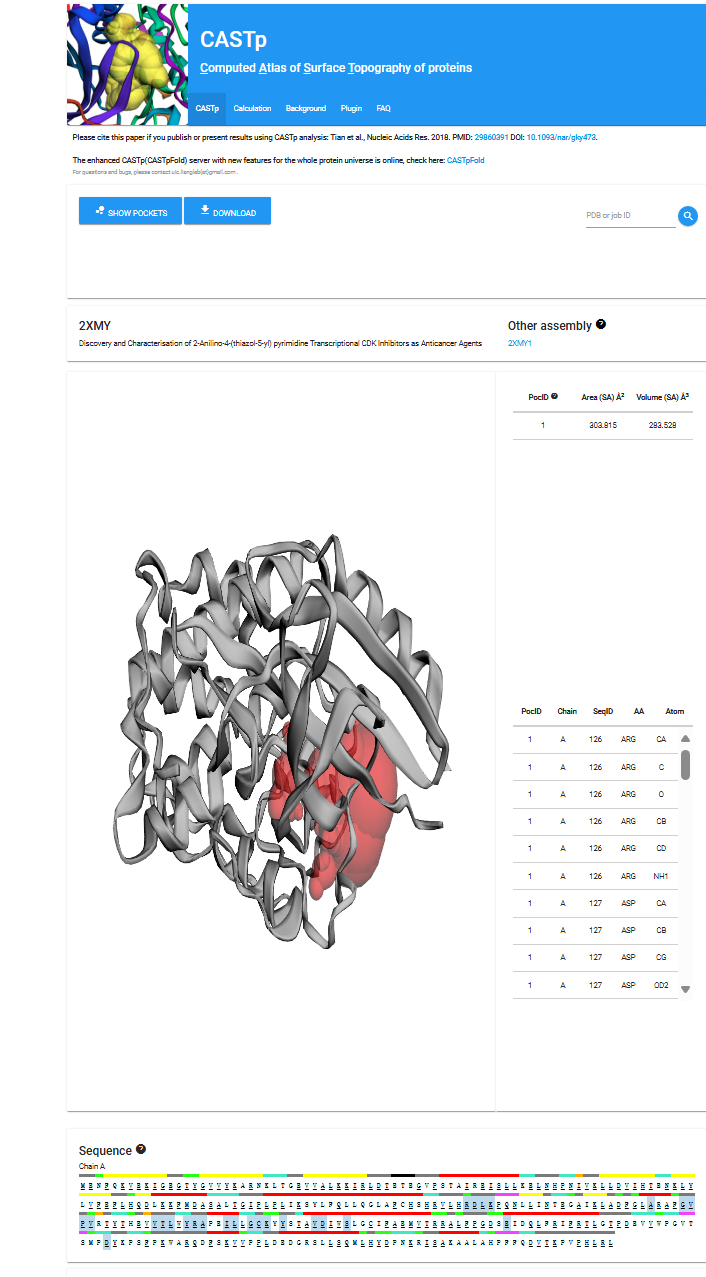
优点:CASTp的核心优势在于其精确、全面且用户友好的口袋分析能力。其采用的拓扑学方法能比传统几何方法更准确地描绘复杂口袋的形状和边界,提供高度可靠的体积和表面积计算结果。
该网站不仅提供口袋的3D可视化(可直接在浏览器中交互操作),还提供极其详尽的定量数据(如体积、面积、开口周长、深度、口袋内表面积占比、组成残基列表及接触面积等),并允许用户根据这些参数进行排序和筛选。
此外,CASTp数据库预计算了PDB中几乎所有结构的口袋信息,数据丰富且更新及时,用户无需本地安装即可快速获取结果,并且完全免费公开访问,使其成为结构生物学、生物信息学和药物发现领域不可或缺的研究利器。
FTMap
地址:https://www.vajdalab.org/ftmap
FTMap 网站是一个基于碎片的结合热点识别平台。它是一个计算映射服务器,用户提交蛋白质、DNA 或 RNA 的 PDB 格式结构后,FTMap 会利用 16 种小有机分子作为探针,对数十亿个位置进行采样,并使用详细的能量表达式对探针的位置进行评分。
能识别出大分子的结合热点,即对配体结合自由能有主要贡献的表面区域,确定的结合热点与实验数据吻合良好。FTMap 还为其他服务器提供了基础,如用于预测配体结合位点的 FTSite、考虑侧链灵活性的 FTFlex 等。
优点:准确性高,比传统的网格和 MCS 等映射方法更精确,能更准确地识别结合热点,为后续的研究提供可靠的基础。运算速度较快,对于平均大小的蛋白质,使用 16 种探针分子能在 1 小时内找到其热点,相比基于混合分子动力学的蛋白质映射方法要快得多。
此外,FTMap 网站功能丰富,基于它发展出了一系列相关服务器,可满足不同用户在蛋白质结构分析方面的多种需求,如预测配体结合位点、考虑侧链灵活性等。
HotSpot Wizard
地址:https://loschmidt.chemi.muni.cz/hotspotwizard/
HotSpot Wizard 网站是一个用于蛋白质工程中自动识别 “热点” 以及对蛋白质结构进行注释的网络服务器。
它整合了来自 RCSB PDB、UniProt、PDBSWS、Catalytic Site Atlas 和 nr NCBI 等数据库以及 CASTp、CAVER、BLAST、CD – HIT、MUSCLE 和 Rate4Site 等工具的结构、功能和进化信息,仅需用户输入蛋白质结构和电子邮件地址即可进行计算。
该网站会列出按估计可变性排序的注释残基,分析结果会映射到酶结构上,并可使用 Jmol 在网络浏览器中可视化,为蛋白质工程师探索蛋白质结构以及设计定点诱变和定向进化实验中的突变提供帮助。

优点:高度集成了多种生物信息学数据库和计算工具,能综合多方面信息来准确识别蛋白质工程中的 “热点”,为用户提供全面且准确的分析结果。使用方便,用户只需提供蛋白质结构和邮箱地址这两个必要输入项,即可轻松获得计算结果。
结果呈现方面也很出色,不仅能将分析结果直观地映射到蛋白质结构上并通过 Jmol 在网页浏览器中可视化,还提供了详细的注释信息,包括残基的可变性、结构位置、功能作用等,方便用户深入了解蛋白质的相关特性。此外,该网站自动化程度高,实现了蛋白质工程协议的自动化,降低了使用门槛,使更广泛的科研群体能够受益
P2Rank
地址:https://prankweb.cz
P2Rank 是一款基于机器学习的蛋白质配体结合位点预测工具,可通过官网获取。它是一个独立的命令行程序,也提供 Java 库。
其核心算法是在蛋白质的溶剂可及表面上对位点进行评分和聚类,利用在已知蛋白质 – 配体复合物数据集上训练的机器学习模型,来确定每个点的配体结合能力评分,从而识别出蛋白质中潜在的配体结合位点,输出概率评分和 3D 结构。
它支持 PDB、mmCIF 和 BinaryCIF 等多种输入格式,能在 Linux、macOS 和 Windows 系统上运行。
优点:预测精度高,通过机器学习模型实现了高精度的配体结合位点预测,相比一些同类工具如 Fpocket 等,在预测成功率上有显著优势。支持多种输入格式,能满足不同用户的数据需求。
使用便捷,无需安装,用户直接下载二进制包即可使用,而且是跨平台兼容的,方便不同系统的用户操作。另外,它还提供了 PyMol 可视化脚本,方便用户直观地查看预测结果,有助于后续的分析和研究。
ConSurf
地址:http://consurf.tau.ac.il
ConSurf 网站是一个免费的生物信息学服务器。它主要用于分析蛋白质或核酸分子中氨基酸或核苷酸位点的进化保守性。
用户提交蛋白质或核酸的序列或结构后,服务器会自动收集同源序列,进行多序列比对并构建系统发育树,然后在概率框架内估计每个序列位置的进化速率,将进化保守性程度映射到分子表面,以不同颜色展示每个氨基酸的保守程度,从而帮助用户识别出对结构和功能重要的区域。
优点:自动化程度高,从同源序列收集、多序列比对到进化速率估计等一系列复杂分析过程都可自动完成,无需用户手动干预。结果呈现直观,通过将保守性信息映射到分子表面并用颜色编码,用户能很容易地识别出保守区域和可变区域。
该网站不断更新升级,算法经过同行评审,保证了分析结果的准确性和可靠性。
此外,它还提供了多种新功能,如自动选择最佳进化模型、对查询蛋白质进行同源建模、预测 RNA 分子二级结构等,能满足不同用户的多样化需求。
COACH
地址:http://zhanggroup.org/COACH/
COACH是一个专门用于蛋白质配体结合位点预测的元服务器工具,整合了多种计算方法和数据库资源。用户只需输入蛋白质的三维结构或氨基酸序列,COACH 即可通过 TM-site 和 S-site 两种对比方法从 BioLIP 数据库中识别与目标蛋白结构或序列相似的配体结合模板,同时结合 COFACTOR、FindSite 和 Concavity 等工具的预测结果,最终生成高可信度的结合位点注释。
预测结果不仅包含按置信度排序的结合残基列表,还可通过 Jmol 插件在网页中直接可视化蛋白质 – 配体相互作用的三维结构,并提供结合位点的功能注释信息。若输入为序列,COACH 会先通过 I-TASSER 生成蛋白质三维模型,再进行结合位点预测,适用于无已知结构的蛋白分析。
优点:作为元服务器方法,它融合了多种独立算法的预测结果,显著提升了结合位点识别的准确性和鲁棒性,在 CAMEO 国际盲测中表现优异;支持灵活的输入方式,无论是已解析的三维结构还是原始氨基酸序列都能处理,尤其适合无结构信息的蛋白研究;结果呈现直观且功能丰富。
用户不仅能通过可视化工具观察结合位点的空间分布,还能获取结合残基的功能注释、进化保守性分析等深度信息;此外,其后台集成的 BioLIP 数据库包含大量经过人工校验的蛋白质 – 配体相互作用数据,为预测提供了可靠的模板支持。
对于需要进一步优化配体结合姿势的用户,COACH 的升级版 COACH-D 还引入分子对接技术,可有效减少预测模型中的空间冲突,生成更接近真实结合模式的复合物结构。