

什么是同步辐射R空间?
精选干货|同步辐射PDF基础知识及经典应用分析!
电解水反应包括阳极的析氧反应(OER)和阴极的析氢反应(HER)。在催化剂服役过程中,其结构和性能会发生动态变化,但目前对这些动态机理的认识仍不充分。
为了深入理解催化剂服役过程中的动态构效关系,原位表征技术结合理论计算分析显得尤为重要。同步辐射技术以其波段宽、亮度高、高偏振性等优势,为电解水催化剂的动态研究提供了强大的工具。从第一代的正负电子对撞机装置到第三代的同步辐射光源,我国的同步辐射技术不断发展。
基于同步辐射的X射线谱学技术,如X射线吸收谱(XAS)、X射线衍射谱(XRD)和X射线光电子能谱(XPS),为电解水催化剂的动态结构研究提供了无限可能。
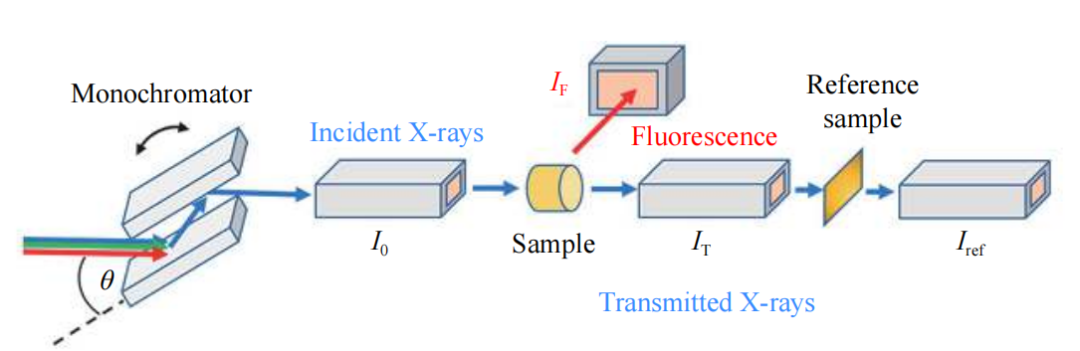
XAS的测量模式主要包括透射模式和荧光模式,研究者可以根据样品的具体情况选择合适的测量方式。
透射模式对样品的质量要求较高,能够提供高质量的谱图数据,而荧光模式则在探测低掺杂量样品时表现出色。原位XAS技术能够在真实的反应环境下实时监测选定元素的氧化态和配位环境的动态变化。
值得注意的是,XAS所获取的是材料整体的平均信息。然而,催化反应主要发生在材料表面,对于大块样品而言,表面元素的信息可能会被体相信息所掩盖,从而给催化研究带来一定的挑战。
目前,有两种方法可以解决这一问题:一是将催化剂制备成纳米尺寸,这样可以确保表面信息不会被体相信息所中和;二是当研究的元素仅分布在材料表面时,此时所得到的元素信号即代表了表面的信号。
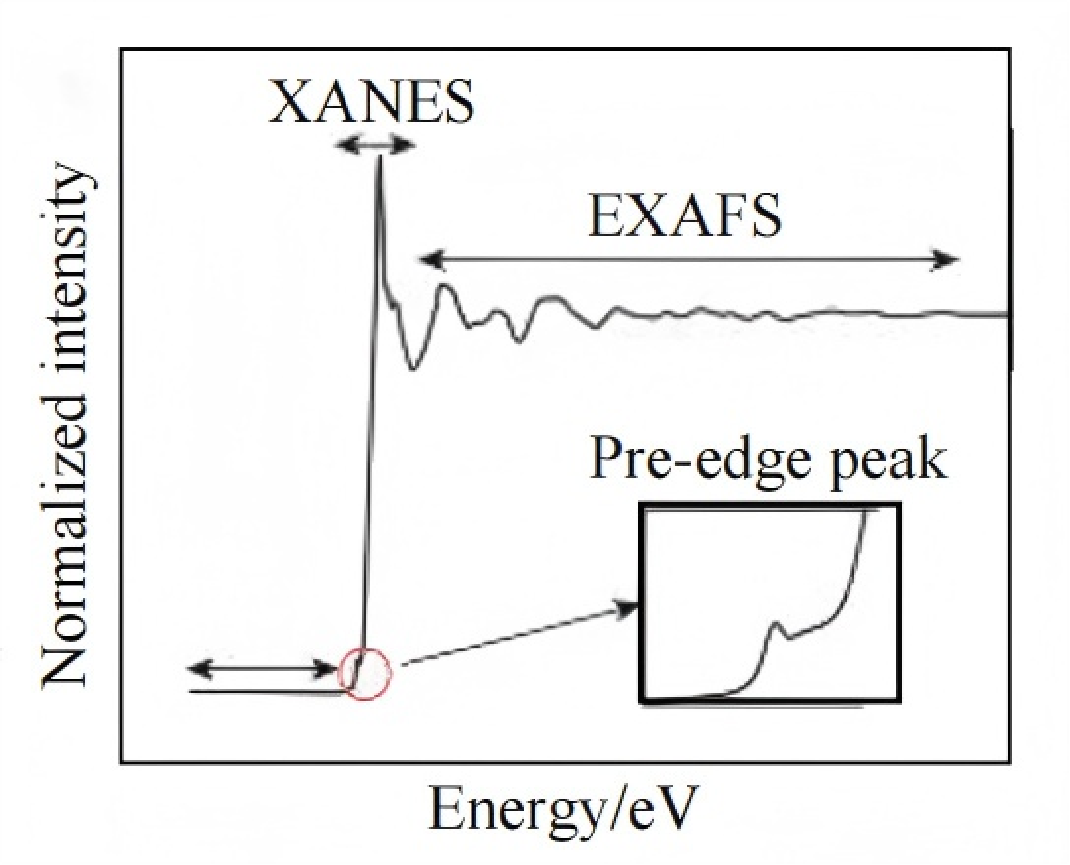
XAS主要由两部分组成:X射线吸收近边结构谱(XANES)和扩展边X射线吸收精细结构谱(EXAFS),这两部分分别包含了丰富的结构信息。常规的XAS装置通过单色器对入射X射线的能量进行筛选。
透射模式通过比较入射X射线强度I₀和透过样品后的强度Iₜ来获取样品信息,而荧光模式则是通过比较I₀与样品反射的X射线强度Iₕ。最后,参比样品Iₓ用于数据校正。

XANES
位于谱图吸收峰附近,能够提供吸收原子的氧化态和局部对称性信息。其原理是原子吸收X射线后,内层电子向外层跃迁。
紧随其后的是EXAFS区域,该区域的振荡反映了吸收原子产生的电子波与近邻配位原子之间的相互作用,其范围可延伸至约1000 eV。
与XANES相比,EXAFS受电子结构细节的影响较小,主要受吸收原子最近邻原子的空间排列影响,因此可以用来表征所选元素的配位数、键长和无序度等信息。
由此可见,XANES和EXAFS这两部分为材料结构提供了高度互补的信息,有助于全面理解催化材料在服役过程中的动态行为。
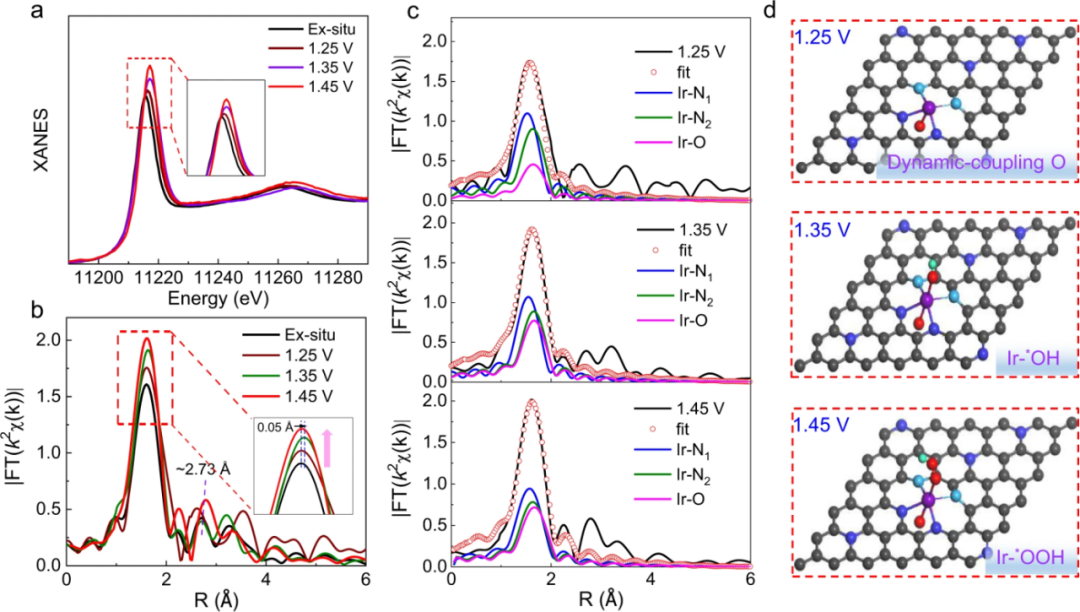
析氧反应(OER)涉及多电子转移过程,动力学反应较慢,揭示其催化反应机理对于提高电催化剂活性、稳定性和降低能源转换成本至关重要。中国科学技术大学韦世强团队借助原位XAFS实验技术从原子级别表征了Ir单原子催化剂在电催化析氧反应过程中的真实活性结构及其动态演变。
他们通过电驱动氨基沉积策略开发了异氮配位的Ir单原子催化剂(AD-HN-Ir)并用于催化酸性OER。原位XANES表征结果如图所示,在反应电位驱动下电子从Ir位点向周围配位原子转移,金属Ir的电子空位增加。
图中的EXAFS图谱显示,在预催化阶段的1.25 V电位下,非金属配位壳层(Ir-N/O)的峰值强度与非原位状态相比有所增强。相应的XAFS拟合分析显示,除了初始的Ir-N4构型外,Ir位点上出现一个与氧吸附有关的Ir-O键(图c)。
当电位升高到1.35和1.45 V时,Ir–N/O配位峰的强度进一步增强,扩展边中相应的峰位置出现偏移,这可能与吸附在单位点上的羟基进一步演化形成的*OOH中间体有关。该原位表征深入解析了O-hetero-Ir-N4单原子活性位点在催化OER的动态作用过程。
DOI:10.1038/s41467-021-26416-3
X 射线衍射(XRD)是为最常见的表征材料晶体结构的技术手段。基本原理为布拉格公式:2dsinθ=nλ。式中,d 为两个晶面之间的间距 , θ 和 λ 分 别 为X 射线束的入射角和波长,n 是整数。
通过测量入射 X 射线的角度和衍射 X 射线的强度,最后绘制出强度与散射(2θ )的关系曲线,结合 PDF 卡片就可以确定样品的晶体结构。
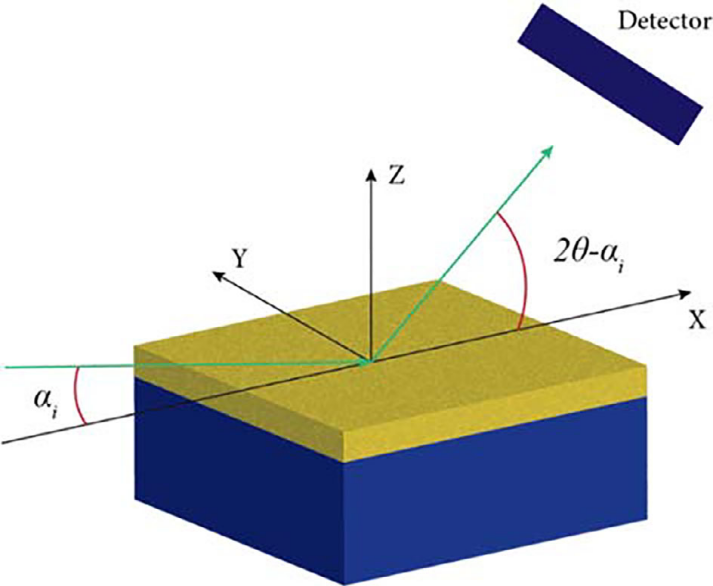
掠入射X射线衍射(Grazing – Incidence XRD,简称GIXRD)是一种表面敏感技术,样品被放置成使样品表面与入射X射线束之间形成一个小角度(通常在1°到3°之间)。
下图展示了GIXRD数据检测的示意图。表面X射线衍射(Surface XRD)技术用于研究材料表面结构、晶区与材料主体结构不同的界面,以及表面现象。
基于同步辐射的X 射线衍射装置可以设计反应池来进行原位XRD 表征,用来探究晶体结构和微晶粒径的演变,对于那些在反应过程中整体结构发生变化的体系,原位 XRD 具有较好的效果。
对于纳米尺寸的薄膜样品,往往普通 XRD 的信号很弱,而掠入射XRD 可以用来增强材料表面的结构信息。
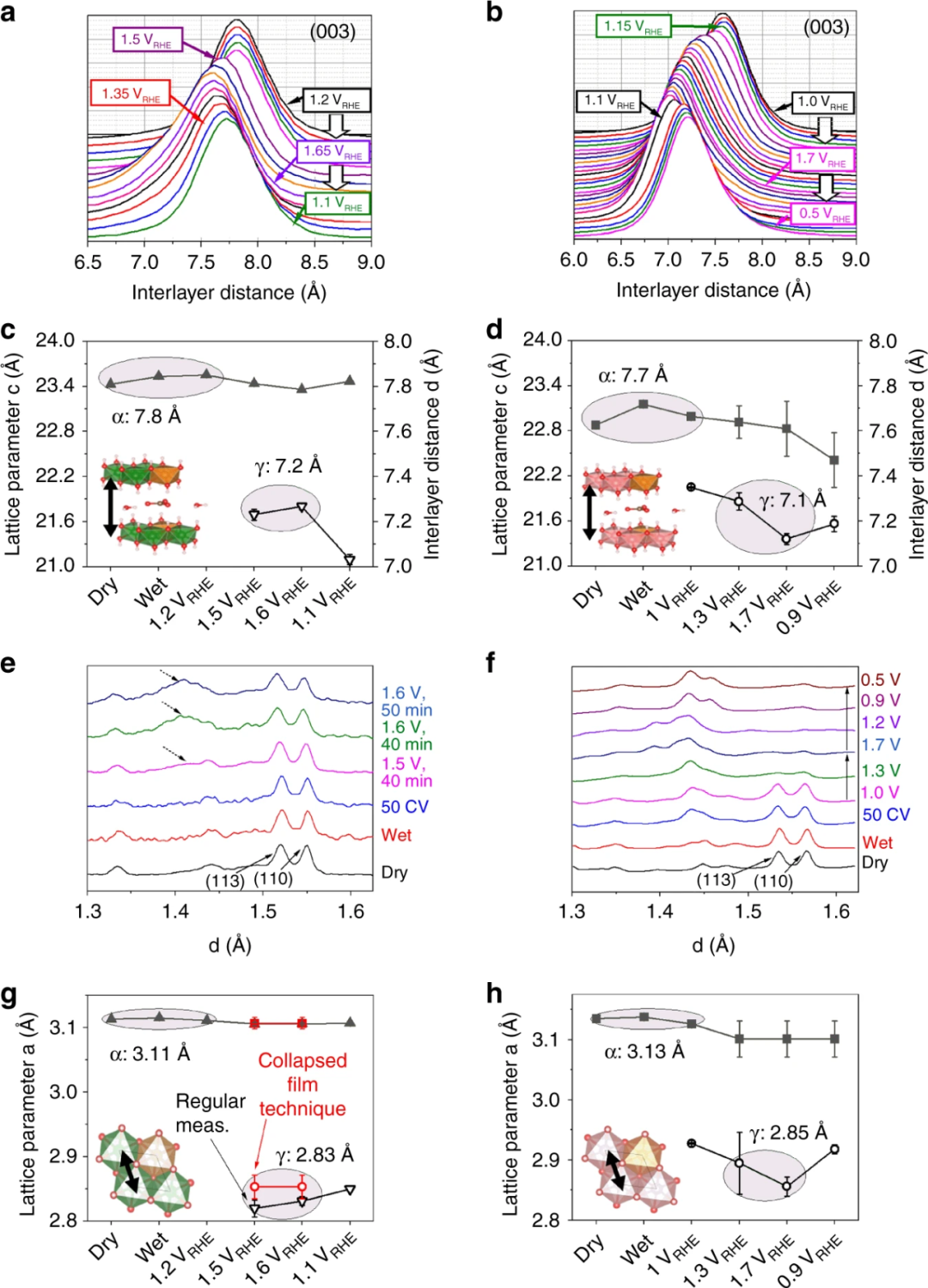
柏林工业大学 Dionigi 团队利用基于同步加速器的动态现场原位广角X射线散射(WAXS)技术,从原子级别追踪了NiFe和CoFe层状双氢氧化物(LDHs)催化剂在电催化析氧反应(OER)过程中的结构转变及其动态演变。
在OER条件下,NiFe和CoFe LDHs从合成的前驱体α相转变为活性γ相。具体而言,α相的层间距离分别为7.8 Å(NiFe)和7.7 Å(CoFe),类似于α-Ni(OH)₂结构。
当电势增加到M(II)氧化电势以上时,(003)衍射峰表明层间距离收缩至7.2 Å(NiFe)和7.1 Å(CoFe),接近含水的γ-NiOOH相,因此被称为γ-MFe LDH。在阴极扫描期间,随着还原为M(II),层间距离开始重新膨胀,但CoFe LDH的重新膨胀不完全。
此外,(110)衍射峰显示,从α到γ相变时,金属间距从3.1 Å缩短到2.85 Å,表明面内键的收缩。
这些结果表明,在OER条件下,NiFe和CoFe LDHs的层间距离和面内键均发生显著收缩,从而揭示了其催化活性相的真实结构及其在反应过程中的动态演变。
DOI:10.1038/s41467-020-16237-1
当X射线束的能量高于样品的功函数时,入射的X射线光子会被样品原子内部的电子吸收,从而引发光电子的发射。通过测量这些射出光电子的动能,并结合已知的入射X射线能量,我们可以计算出电子的结合能。
最终,我们可以得到光电子强度与结合能之间的关系图,这就是X射线光电子能谱(XPS)。XPS不仅可以识别样品的元素组成,还可以确定元素的氧化态。
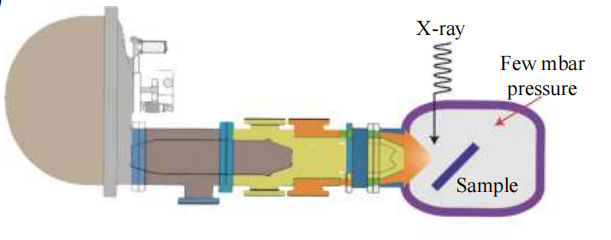
然而,由于光电子在常压下的非弹性平均自由程较短,XPS通常需要在超高真空环境中进行,这使得固液界面的原位电化学分析变得极为困难。为了实现对电极–电解质界面的原位追踪,自20世纪70年代起,研究人员就开始探索近常压条件下的XPS技术。
最初,这种技术主要依赖于实验室的X射线光源。然而,随着2000年后同步辐射装置的迅速普及,近常压X射线光电子能谱(APXPS)的发展迎来了新的机遇。
同步辐射源能够提供高亮度的X射线,并且其能量可以在较宽的范围内自由调节,这为XPS的表面研究提供了更大的灵活性。
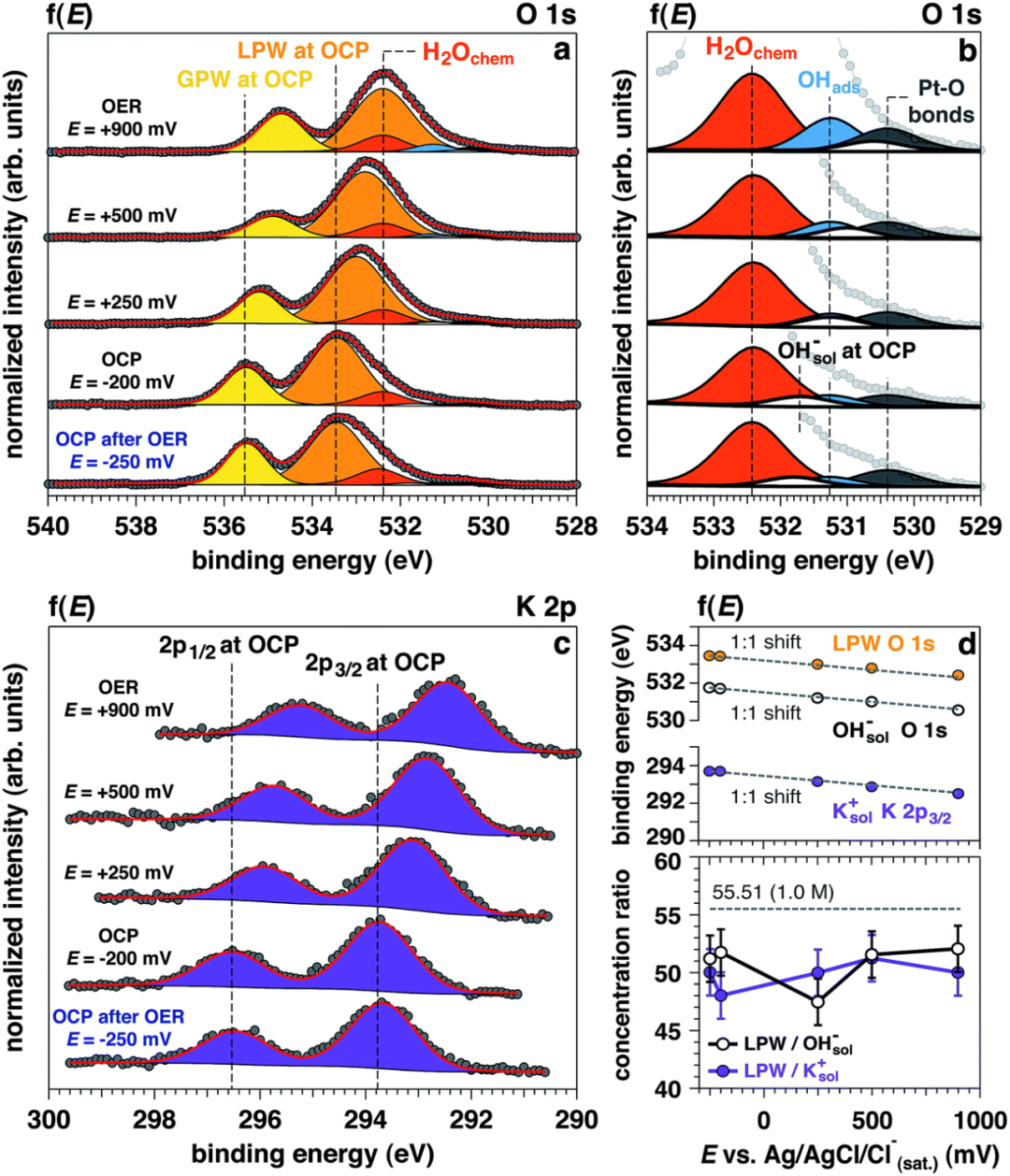
天津理工大学鲁统部团队为了深入理解铂(Pt)电极在碱性条件下氧进化反应(OER)中的表面化学变化,利用原位环境压力X射线光电子能谱(APXPS)技术,揭示了多晶铂电极在1.0 M KOH电解液中氧进化反应(OER)条件下的表面化学变化。
实验中,O 1s 和 K 2p 的光电子峰面积保持稳定,表明溶液中氢氧化物离子(OHsol⁻)与钾离子(K⁺)的浓度比维持在51.4±2.1,接近理论值。
随着施加电位的增加,Pt-O(530.4 eV)和吸附氢氧根(OHabs⁻,531.3 eV)的峰面积显著增加,而在OER条件(+900 mV)下,化学吸附水(H2Ochem,532.4 eV)的峰面积也有所上升。
这些结果表明,在OER条件下,铂表面的氧化态发生了显著变化,形成了更高氧化态的Pt(II)和Pt(IV)物种。
DOI:10.1039/C7TA00409E
先进的同步辐射原位X射线技术,如XAS、XRD和XPS,已成为研究催化材料在实际工作条件下动态行为的关键工具。这些技术不仅帮助研究者深入理解了催化材料的结构和性能变化,还为设计新型高效催化材料提供了重要依据。
尽管如此,这些技术仍存在局限性,例如XAS只能提供整体平均信息,XRD对样品条件要求较高,而XPS的高真空需求限制了其在复杂环境下的应用。
随着新一代同步辐射光源的不断发展,有望突破这些限制,实现对催化材料全生命周期的动态构效关系的精确研究,推动催化科学向更精细化和可控化的方向发展。