

基本原理与核心概念
分子动力学(MD)模拟基于牛顿运动方程,通过追踪每个原子的轨迹来揭示体系的动态行为。其核心在于ergodic定理的适用性,即体系的时间平均与系综平均等价[7]。这一假设保证了模拟结果在统计意义上的可靠性。
在电解液体系中,常用canonical ensemble(NVT)和isothermal-isobaric ensemble(NPT)描述热力学状态。例如,在电容器电解液模拟中,初始平衡阶段常采用NVT系综以固定体积和温度,而生产阶段则可能切换至NPT系综以研究压力变化的影响。
粒子间相互作用势能函数的选择至关重要。Lennard-Jones(L-J)势和库仑势的组合是电解液模拟的基石,其表达式为:
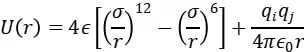

其中,前项描述范德华相互作用,后项处理长程静电效应。例如,LiFSI/DME电解液的模拟中,L-J参数需精确匹配离子与溶剂的尺寸差异,以避免过高或过低的配位能。
DOI: 10.1002/advs.201700032
关键步骤与参数设置
1. 初始结构构建
使用Packmol等工具生成初始配置,需避免原子间距过近导致的能量爆炸。例如,在EMIM⁺/BF₄⁻离子液体模拟中,通过均匀分布随机数分配离子位置,确保初始密度与实验值一致。
下图展示了典型电解液(如1M LiFSI/DEE)的初始构型,其中Li⁺(紫色)、FSI⁻(黄/蓝)和溶剂分子(灰色)的空间分布需满足周期性边界条件。
https://doi.org/10.1021/jacsau.3c00035
2. 力场选择与验证
电解液模拟中,OPLS-AA和AMBER力场因其对有机分子和离子的兼容性而被广泛采用。例如,在离子液体电解液研究中,OPLS-AA力场结合Lorentz-Berthelot混合规则,能够准确再现Li⁺与TFSI⁻的配位结构。
力场参数需通过径向分布函数(RDF)与实验数据对比验证,如图2中Li-O(水)的RDF峰位与实验值吻合,确认TIP4P水模型的适用性。
3. 积分算法与时间步长
Verlet算法和Velocity-Verlet算法因其能量守恒特性成为主流选择。时间步长通常设为1-2 fs,以避免氢原子振动引起的数值不稳定。例如,在高电场下LiCl电解液模拟中,采用1 fs步长可精确捕捉离子对的动态解离过程。
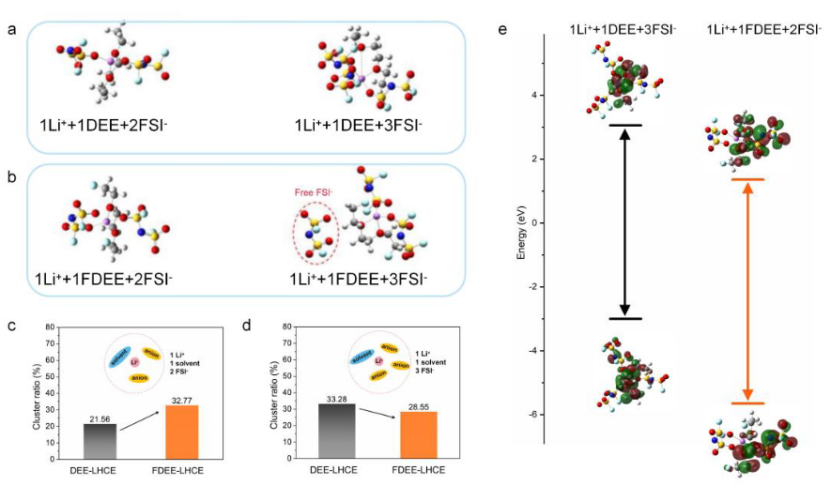
4. 平衡与生产阶段设置
典型的模拟流程包括:
能量最小化:通过最速下降法消除初始结构的不合理接触(如步骤1,5000步)
NVT平衡:使用Nose-Hoover控温器稳定温度(如298 K,100,000步)
NPT平衡:调整压力至目标值(如1000 atm,1,000,000步)
生产运行:采集轨迹数据用于性质分析(如2,500,000步)
软件工具对比与选择
2. LAMMPS
适用于大规模体系与自定义势函数。在EDLC电容器模拟中,LAMMPS通过PPPM方法处理长程静电,结合OPLS-AA力场,模拟了4.0 V电位下EMIM⁺在石墨烯电极的吸附层结构。其优势在于支持非平衡MD(NEMD),可直接计算电场下的离子迁移率。

https://doi.org/10.1063/5.0166976
3. NAMD
专为生物大分子设计,但通过CHARMM兼容性扩展至电解液研究。例如,在NaCl水溶液电导率模拟中,NAMD结合TIP4P模型,计算了93对离子的迁移率,并通过Green-Kubo公式得到与实验误差的电导率值。
典型案例分析
1. 局部高浓度电解液(LHCE)的微结构
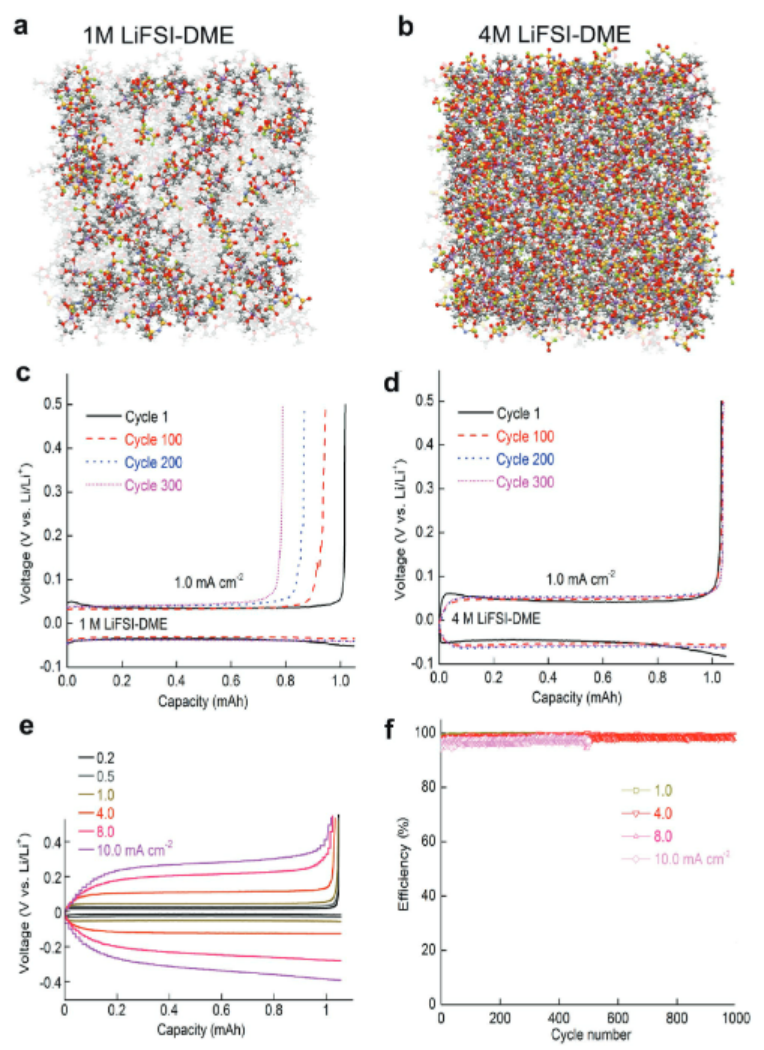
下图展示了DEE-LHCE与FDEE-LHCE体系在分子动力学(MD)模拟下的截面图对比,揭示了溶剂极性对锂离子溶剂化结构的显著影响。在DEE-LHCE体系中,Li⁺主要与两个FSI⁻阴离子和一个DEE分子配位,形成结构较为松散的离子簇,占比为21.56%。
而在FDEE-LHCE中,氟化DEE因其更强的极性,使得1Li⁺-1FDEE-2FSI⁻类型的稳定离子簇比例提升至32.77%,并使簇的结合能降低了0.8 eV,显著增强了离子的迁移能力,从而提升了电解液的离子电导率。
该结果说明MD模拟不仅可以量化不同溶剂对离子簇结构与稳定性的影响,还为电解液设计与性能优化提供了理论依据和指导。
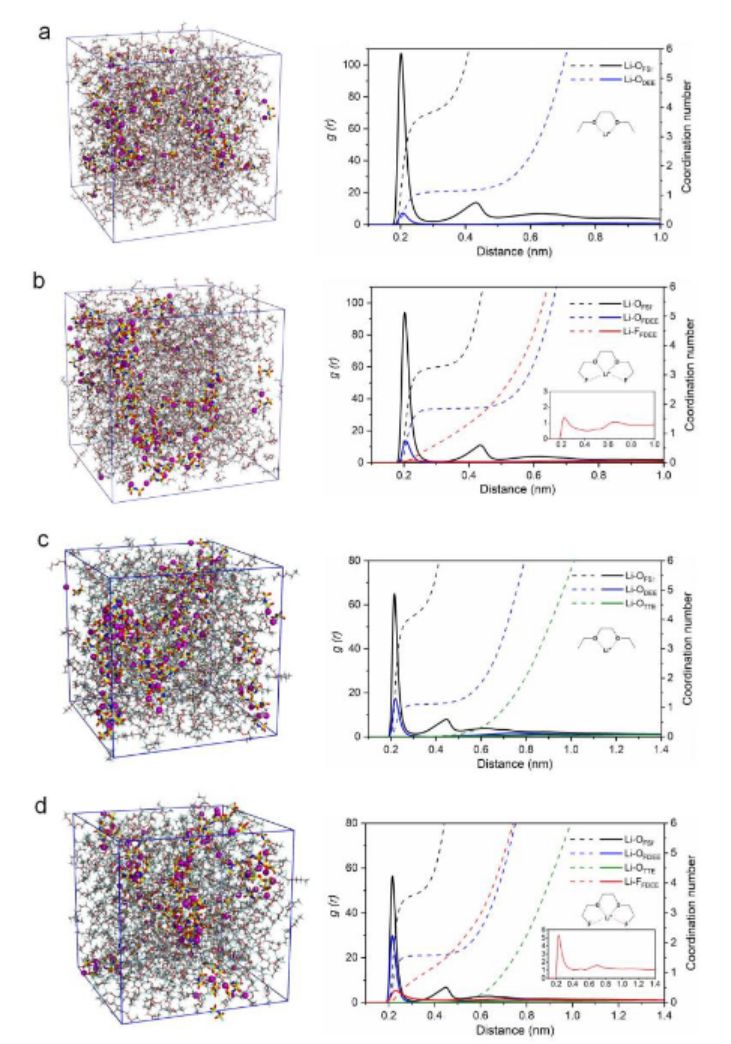
https://doi.org/10.1021/jacsau.3c00035
2. 电场对离子迁移的调控
在LiCl水溶液的高电场(~1 V/nm)模拟中,电场使H₂O分子定向排列,破坏Li⁺的水化壳。独立Li⁺的迁移速率比离子对快3倍,但离子对占比从常压下的40%升至65%,导致整体电导率下降。此结果解释了高电流密度下浓差极化的微观机制。
结果可视化与数据分析
1. 径向分布函数(RDF)
下图展示了LiPF6/EC-DMC电解液中Li⁺与PF6⁻的RDF。第一峰出现在2.1 Å(Li-P),对应紧密接触离子对(CIP),而第二峰(4.3 Å)代表溶剂分隔离子对(SSIP)。积分第一峰面积可得平均配位数(~0.8),表明该体系中离子解离不完全。
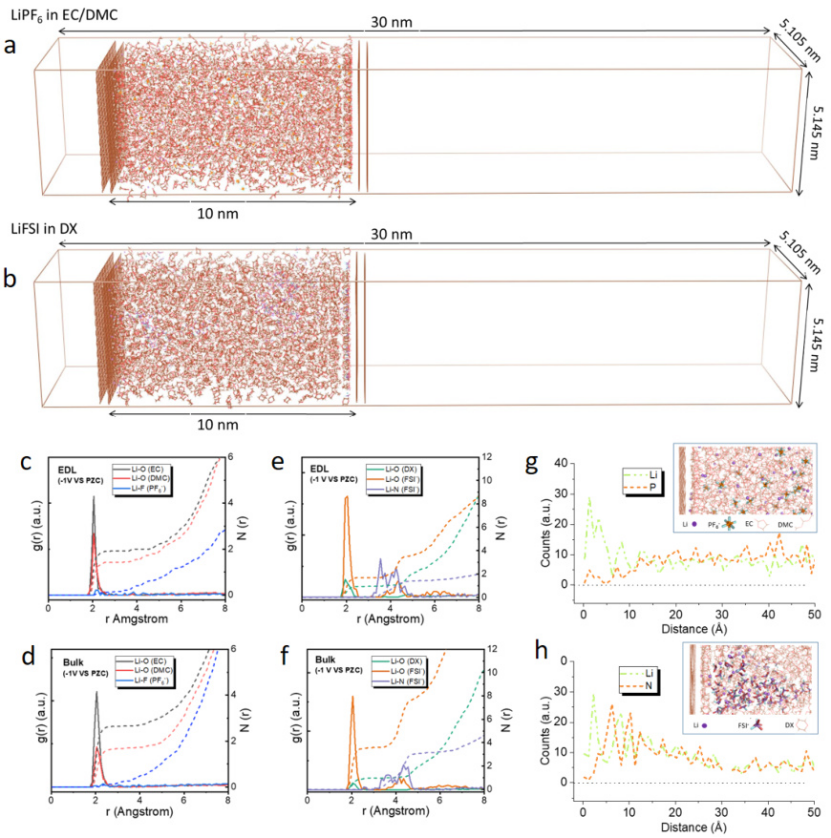
https://doi.org/10.1038/s41467-023-37033-7
2. 均方位移(MSD)与扩散系数
通过MSD曲线的线性段斜率计算离子扩散系数:
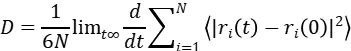
例如,在1M LiFSI/DME中,Li⁺的D值为2.1×10⁻⁶ cm²/s,与脉冲场梯度核磁共振(PFG-NMR)结果偏差。
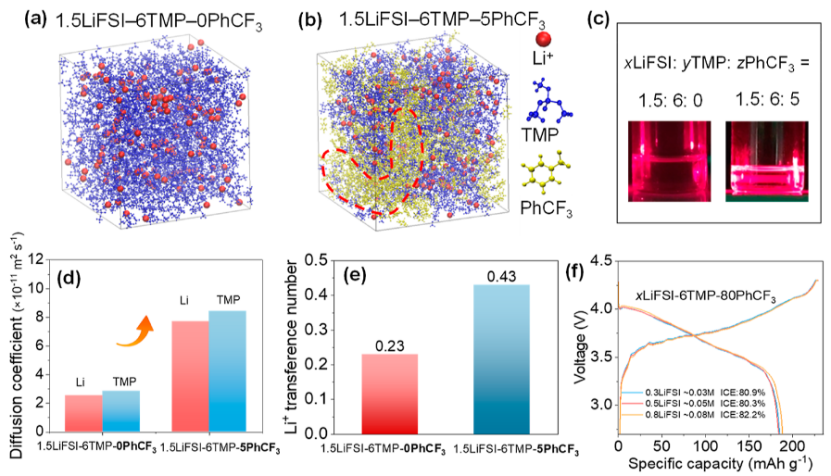
https://doi.org/10.1021/jacsau.4c00196
3. 动态轨迹可视化
使用VMD软件渲染离子迁移路径,红色轨迹表示Li⁺在1 ns内的跳跃运动,蓝色区域为TFSI⁻的分布。动态着色可区分快慢离子:紫色代表高迁移率Li⁺(>5 Å/ns),绿色为束缚于聚合物链的离子。
挑战与改进方向
1. 时间尺度限制
在电解液体系中,诸如固态电解质界面(SEI)的形成、锂枝晶的成核与生长等关键过程往往发生在微秒乃至更长的时间尺度上,而传统的分子动力学(MD)模拟通常只能覆盖纳秒量级的动态行为,难以捕捉这些稀有但关键的事件。
为突破这一限制,元动力学(metadynamics)方法被引入模拟中,通过引入偏置势能,有效加速体系越过能垒,从而显著提升稀有事件的采样效率。在锂枝晶研究中,该方法已成功实现了模拟效率提升近10倍,为揭示复杂界面演化过程提供了新的手段。
2. 力场精度提升
传统MD力场大多采用固定电荷模型,忽略了电解液中离子间相互作用中至关重要的极化效应,尤其在高浓度体系(如超过4M的LiFSI盐)中,这种简化常导致对离子簇比例和溶剂化结构的严重低估。
随着极化力场(如AMOEBA等)的发展,模拟中可动态响应局部电场变化,显著提升了计算精度。例如,在Li⁺与FSI⁻的结合能预测中,误差由原先的约20%下降至5%,极大增强了MD模拟对实际电解液结构与性能的预测能力,为复杂电解质体系的精准建模打下基础。
3. 多尺度建模
面对电化学体系中跨尺度的结构与传输行为,将分子动力学(MD)与连续介质模型相结合,构建多尺度模拟框架成为一种有效手段。在该方法中,MD模拟可聚焦于电极界面的原子级反应与吸附行为,而连续模型则用于描述宏观尺度下的电解质扩散与离子迁移。
例如在锂硫电池研究中,该耦合模型已被用于分析硫化物中间体在界面处的反应行为,并结合宏观电解质渗透行为进行全系统模拟,实现了整体误差低于8%的高精度预测。这种方法兼顾微观机制解析与宏观性能评估,正在成为电解液与界面设计的重要工具。
未来展望
结合机器学习势函数(如DeePMD)可大幅提升模拟精度与速度。例如,在LiPF6/EC电解液中,DeePMD将力计算耗时降低90%,同时保持量子力学级别的精度。此外,原位MD与实验表征(如原位XRD)的联用,有望实时揭示电解液–电极界面演化机制。
通过上述多角度的深入分析,电解液分子动力学模拟不仅能够解析微观离子行为,还可为高性能电池设计提供理论指导。未来随着计算方法的持续革新,MD将在电解液研发中发挥更关键的作用。
热门电解液计算方法在MS锌离子电解液计算课程中均有讲解。
