

在分子动力学(MD)模拟中,氢键统计通常基于几何准则进行判断:当供体原子(如O-H)与受体原子(如O或N)之间的距离小于设定阈值(一般为0.30–0.35 nm),且供体—氢—受体之间的夹角大于某个角度(通常>120°或氢键。
模拟过程中,每隔一定步数(如每1 ps)记录一次氢键数量,并生成随时间变化的轨迹。
进一步可通过计算氢键的生存率(存在时间占比)、平均数量或绘制热图与径向分布函数(RDF),来分析氢键网络的稳定性、动态特征及关键相互作用位点。
这种方法可以直观揭示体系内部微观结构和分子识别机制。
氢键的定义与基本特性
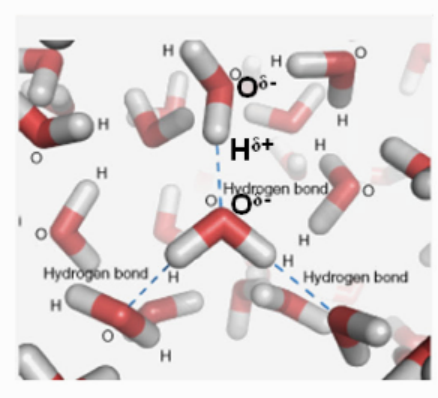
氢键是一种介于化学键与范德华力之间的特殊相互作用力,其本质是电负性较大的原子(如O、N、F)通过氢原子媒介形成的弱键合作用。
具体来说,当氢原子与高电负性原子X(如O)以共价键结合时,由于X对电子的强吸引力,氢原子呈现部分正电荷(δ+);此时另一个电负性大的原子Y(带有孤对电子)会被吸引,形成X—H···Y的三中心相互作用。
这种作用力具有以下特点:
强度范围:约为5-40 kJ/mol,显著强于范德华力(5 kJ/mol),但远弱于共价键(150 kJ/mol)。
方向性:X—H···Y的键角通常接近180°,以减小X与Y之间的电子云排斥。
饱和性:一个H原子仅能与一个Y原子形成有效氢键,因其体积小且电荷有限。
反转电性氢键:近年研究发现,当X的电负性小于H时(如Si—H),可能形成“反转电性氢键”(Xδ+—Hδ−···Yδ−),例如在三甲基硅烷中,H的负电性吸引路易斯酸。
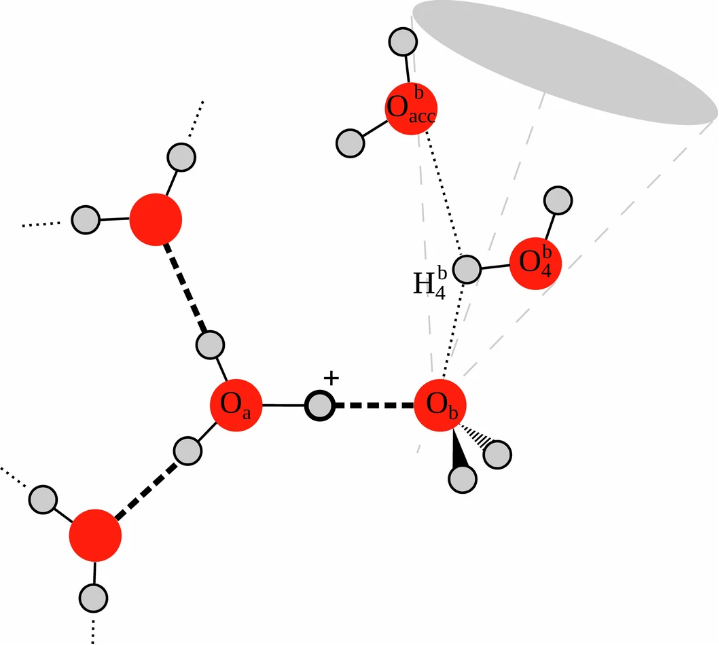

(Nat. Chem. 16, 1838–1844 (2024). https://doi.org/10.1038/s41557-024-01593-y)
氢键的判断标准
在分子动力学(MD)模拟中,氢键的识别依赖于几何准则或能量准则,其中几何准则因计算效率高而被广泛应用。
不同软件和文献中采用的具体参数略有差异,需注意标准化设定:
几何准则的核心参数
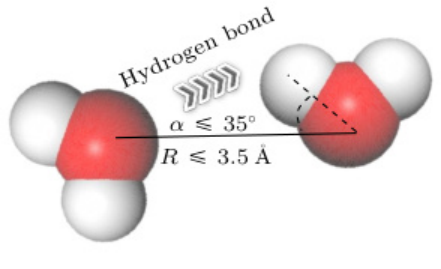
距离阈值:施体(X)与受体(Y)的原子间距(R)通常≤3.5 Å(0.35 nm)。例如,水分子间O···O距离≤3.5 Å时视为氢键形成。
(http://doi.org/10.7498/aps.73.20240192)
角度阈值:X—H···Y的键角(θ)需≤30°(部分文献定义为施体–受体–氢的夹角≥120°)。
例如,在GROMACS中默认角度阈值为30°,而VMD的HBonds插件采用更严格的20°。
不同软件的实现差异
在不同的软件平台上,氢键的识别和计算存在明显的实现差异,这直接影响模拟结果的准确性和可比性。
比如在GROMACS中,默认采用的是混合的距离–角度准则,通常设定氢键距离阈值为0.35 nm,角度小于30°。
不过,用户也可以根据体系特点,通过调整参数,仅使用距离作为判断标准,从而适配不同分子环境下氢键的定义需求。
相比之下,VMD的默认设置更为严格,采用了3.0 Å(即0.30 nm)的距离限制和更小的20°角度限制,而且默认忽略了周期性边界条件(PBC)。
这在处理液态水等强烈依赖PBC的体系时,可能导致统计到的氢键数目显著低估。
对于一些特殊体系,例如涉及弱氢键相互作用(如C—H···O型氢键),单纯依赖标准阈值可能无法准确捕捉。
此时可以将距离阈值适当放宽至0.40 nm,同时结合能量判据(如结合能低于一定阈值)作为辅助标准,提升识别的合理性和全面性。
这些细节设置对最终分析结果的可靠性有着直接而深远的影响,因此在具体研究中,氢键识别参数的合理选择必须结合体系特性与科学问题目标灵活调整。
(http://commons.wikimedia.org/)
动态分析与生存率计算
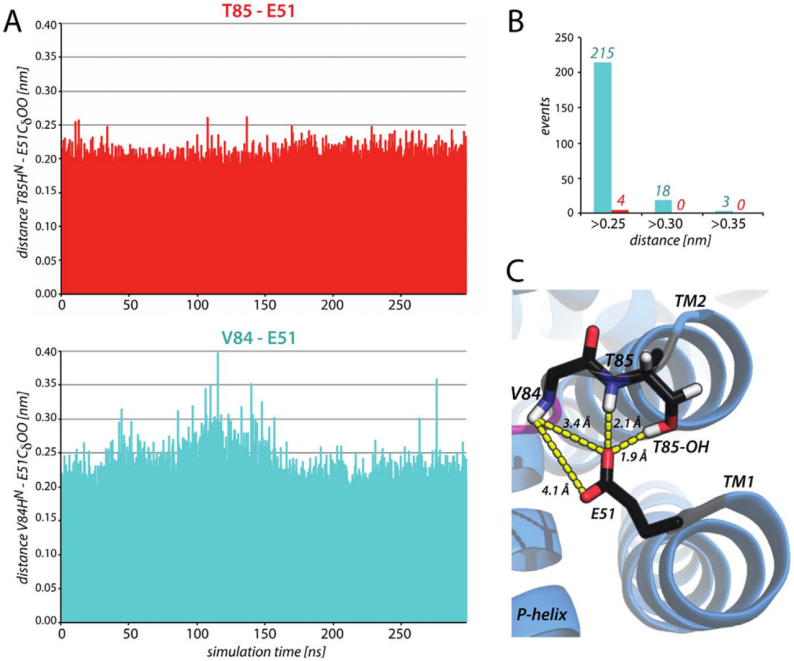
氢键的稳定性常通过生存率(Survival Rate)评估,即某一氢键在模拟轨迹中持续存在的时长占比。
例如,在血小板信号通路研究中,通过计算复合物界面氢键的生存率(Pi)和解离概率(DP=∑(1-Pi)),量化分子间结合的动态稳定性。
(https://doi.org/10.1016/j.ssnmr.2017.03.003)
氢键分析在分子动力学中的作用:以药物设计为例
案例背景:抗COVID-19药物虚拟筛选
文献《Virtual Screening of Secondary Metabolites from Abuta (Cissampelos pareira Linn) as PotentialCOVID-19 Inhibitors: A Computational Approach to Drug Discovery》(Amin et al., 2024)通过分子对接和MD模拟,评估植物化合物Cissampeline与SARS-CoV-2蛋白酶(5R7Y受体)的结合稳定性。

氢键分析的关键作用
在分子模拟与药物设计中,氢键分析起到了至关重要的作用,特别是在结合模式预测与稳定性评估方面。
比如在分子对接阶段,初步结果可能显示Cissampeline与靶蛋白的Val:148位点形成氢键,但通过后续的分子动力学(MD)模拟,可以进一步观察到Cissampeline在动态过程中还与Glu:166、Gly:143等多个位点形成氢键网络。
这表明模拟不仅能够验证静态对接结果,还能捕捉到瞬时产生和断裂的氢键相互作用,提供更加全面的结合模式信息。
另一方面,氢键数目的多少和氢键寿命(生存率)可以作为评估复合物稳定性的重要指标。
通过比较Native ligand、Molnupiravir和Cissampeline的模拟结果发现,Cissampeline在整个轨迹中能形成3-4个氢键,而Molnupiravir通常只有0-1个,而且Cissampeline氢键的生存率也明显更高,说明其结合界面更加稳定。
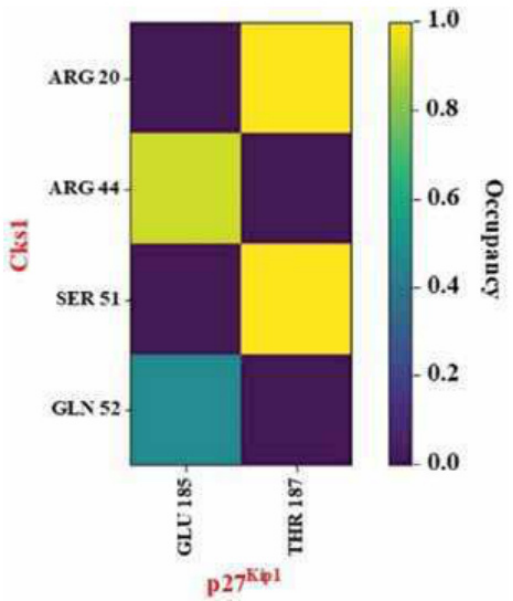
这些信息通常通过氢键热图直观展示,有助于筛选潜在高效的候选分子。
因此,氢键分析在揭示分子识别机制、优化分子结构和指导实验验证方面具有不可替代的价值。
方法学启示
在氢键研究中,方法学上的提升极大丰富了数据的深度与可靠性。
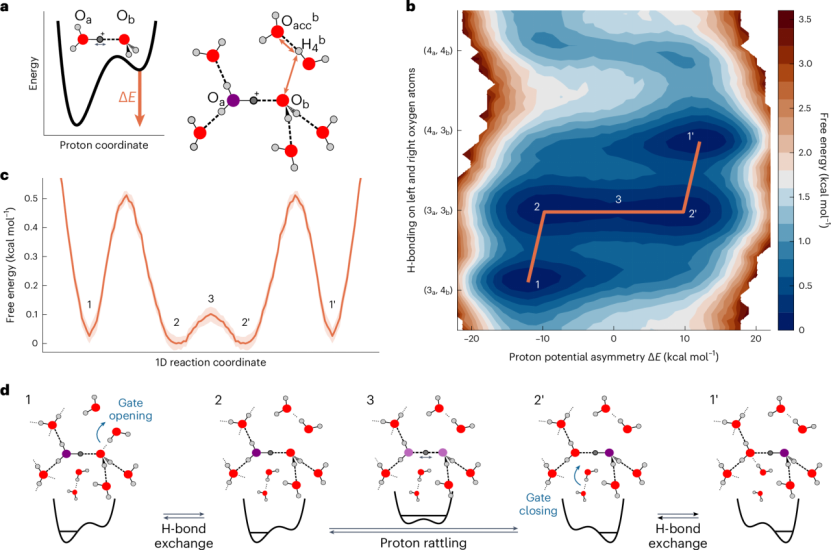
首先,多尺度验证成为了一种重要策略。通过结合量子化学计算(如QTAIM分析)与经典MD模拟,不仅可以从宏观尺度上观察氢键的形成与断裂,还能在电子尺度上精确量化氢键的电子密度分布、拉普拉斯值及能量贡献。
这种跨尺度的分析方法,有助于揭示哪些氢键是真正稳定且对分子识别至关重要的,而不仅仅是基于几何准则的统计。
其次,动态轨迹分析为理解氢键网络的演变提供了强有力的工具。通过绘制氢键热图可以直观展示每一对氢键随时间的形成与断裂频率,而径向分布函数(RDF)则进一步揭示了特定残基或基团之间的距离分布特性。
结合这两种分析,不仅可以确定关键残基在不同时间尺度下的协同作用,还能识别那些在短时间内频繁形成断裂但在整体上贡献重要结合能的动态氢键网络,从而为药物设计和分子改造提供更精准的指导依据。
(Nat. Chem. 16, 1838–1844 (2024). https://doi.org/10.1038/s41557-024-01593-y)
总结与展望
氢键的精确分析是分子动力学(MD)模拟中的核心环节,尤其在生物大分子相互作用、药物设计和功能材料研究中扮演着至关重要的角色。
随着研究不断深入,未来氢键分析呈现出几个明显的发展趋势。
首先,多准则融合将成为主流,不仅考虑传统的几何参数(如距离和角度),还会引入能量指标与电子密度拓扑分析(如QTAIM方法),从而更准确地判定真实有效的氢键。
其次,量子效应的整合也越来越受到重视。路径积分分子动力学(PIMD)能够模拟核量子效应,尤其是质子隧穿现象,这对于理解低温条件下氢键网络的异常行为或极端环境下的分子稳定性至关重要。
再次,随着模拟体系规模的扩大,自动化、智能化的氢键分析工具亟需发展。针对动态复杂的体系(如离子液体–水混合物或生物膜环境),开发可以适应多尺度、多团簇结构变化的算法成为重要方向。
综合来看,氢键不仅仅是单纯的分子间吸引力,它在维持结构稳定、调控反应动力学和决定功能特性方面发挥着桥梁作用。
未来,氢键研究将继续促进化学、生物学和材料科学等多领域的交叉融合与创新应用。