

在微观世界中,分子的动态行为如同一场精妙的舞蹈,深刻影响着物质的性质和功能。分子动力学模拟(Molecular Dynamics, MD)作为探索分子尺度奥秘的强大工具,是一种基于经典力学原理的计算方法,通过求解系统中粒子的牛顿运动方程来模拟其随时间的演化行为,在生物、材料、化学等众多领域发挥着关键作用。以下将从原理、基本步骤、软件对比及使用方法等方面进行详细阐述。

分子动力学模拟的原理
理论基础
MD 模拟的根基深深扎根于经典力学之中。在微观层面,粒子间的相互作用错综复杂,而势函数则成为了描述这种相互作用的有效手段。常见的势函数包括 Lennard-Jones 势、Morse 势等。
以 Lennard-Jones 势为例,它通过数学公式精准地描述了分子间的范德华力,既包含了分子相互吸引的长程力部分,也涵盖了分子相互排斥的短程力部分,为模拟分子间的相互作用提供了坚实的理论依据。
系统的总能量构成是理解分子行为的关键。它主要由键合作用和非键合作用两大部分组成。键合作用涉及到键长、键角、二面角等因素,这些因素决定了分子的基本骨架结构,就像建筑物的框架一样,支撑着分子的整体形态。
键合作用则包含范德华力和静电力,范德华力是分子间普遍存在的微弱相互作用力,而静电力则在带电粒子或具有偶极矩的分子间发挥重要作用,二者共同影响着分子间的聚集、分离等行为。
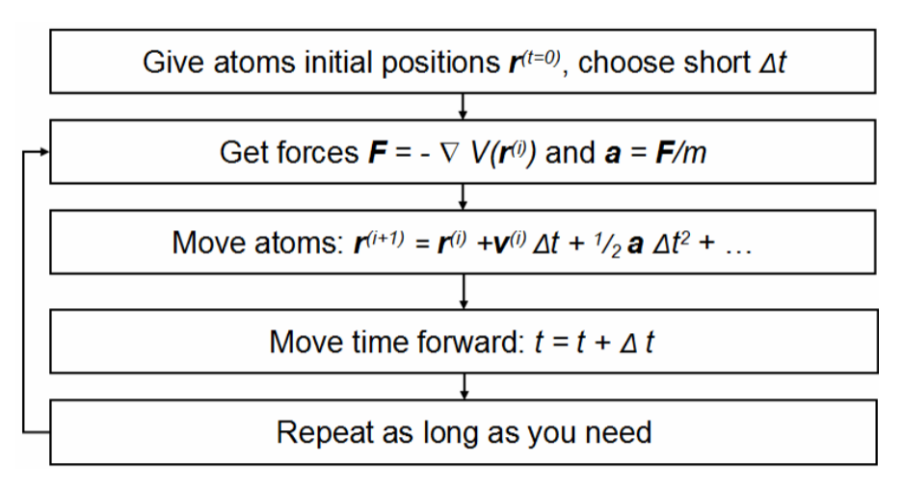
在 MD 模拟过程中,积分算法起着至关重要的作用,常用的 Verlet 算法或 Leapfrog 算法用于更新原子的位置和速度。由于分子运动极其迅速,为了准确捕捉其动态变化并避免数值不稳定,时间步长通常设置为飞秒(10⁻¹⁵秒)量级。
这就好比用高速摄像机以极短的时间间隔拍摄分子运动的画面,从而能够细致地观察分子在每个瞬间的状态。
统计系综是模拟过程中设定环境条件的重要概念。模拟常在 NVE(微正则)、NVT(等温)或 NPT(等压)系综下进行。在 NVT 系综中,为了维持恒定的温度,需要借助恒温器(如 Nose-Hoover 恒温器),它通过调节系统与外界的能量交换,使系统温度保持稳定;在 NPT 系综中,压力控制器则发挥作用,确保系统压力恒定,从而模拟真实的物理环境。
局限性
尽管 MD 模拟在分子研究中取得了显著成果,但它也存在一定的局限性。受限于时间和空间尺度,目前 MD 模拟通常只能在纳米级和纳秒级范围内进行。这意味着对于一些需要长时间、大尺度观察的分子过程,如蛋白质的缓慢折叠过程、材料在长时间作用下的性能变化等,传统 MD 模拟难以胜任。
为了突破这些限制,研究人员不断探索新的方法,如采用粗粒化模型简化分子结构,将多个原子视为一个整体进行模拟,从而减少计算量,扩大模拟的空间尺度;或者运用增强采样技术,提高对分子构象空间的搜索效率,以获取更全面的分子行为信息。
基本模拟步骤
系统初始化
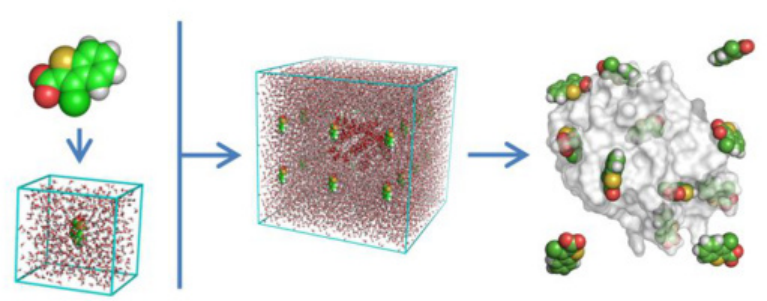
系统初始化是 MD 模拟的起始环节,其中结构构建和力场选择是两个关键步骤。在结构构建方面,可以从晶体结构数据中获取分子的初始构型,许多科学数据库存储了大量晶体结构信息,研究人员能够从中提取所需分子的精确结构。
此外,借助建模软件,如 Materials Studio、VMD 等,也可以灵活地构建复杂分子的三维结构,通过设置原子间的连接方式、键长、键角等参数,生成符合研究需求的分子构型。
力场的选择直接影响模拟结果的准确性和可靠性。不同的力场,如 AMBER、CHARMM、OPLS 等,都有其特定的适用范围。AMBER 力场在生物分子领域表现出色,尤其适用于蛋白质、核酸等生物大分子的模拟。
CHARMM 力场则以其灵活的力场参数设置,广泛应用于生物大分子和膜系统的研究;OPLS 力场对于小分子和溶液体系的模拟具有较好的效果。研究人员需要根据所研究分子的类型和特点,谨慎选择合适的力场,以确保模拟能够真实反映分子的行为。
模拟参数设置
模拟参数设置是为模拟过程创造特定物理环境的重要环节。在物理条件设置方面,温度、压力和溶剂化环境是关键因素。温度决定了分子的热运动剧烈程度,不同的温度条件下,分子的构象变化、反应速率等都会有所不同;压力对一些涉及相变、凝聚等过程的模拟至关重要;
溶剂化环境的选择(显式 / 隐式溶剂)也会影响分子间的相互作用,显式溶剂模型将溶剂分子明确纳入模拟体系,能够更真实地反映溶剂对溶质分子的影响,但计算量较大;隐式溶剂模型则通过经验公式或连续介质模型来近似描述溶剂效应,计算效率较高,但准确性相对较低。
积分参数的设置同样不容忽视。时间步长的选择需要在计算效率和模拟准确性之间进行平衡,通常设置在 0.5–2 fs 之间。时间步长过短会导致计算量大幅增加,而时间步长过长则可能无法准确捕捉分子的快速运动,影响模拟结果的精度。
模拟总时长的确定则取决于研究目的,对于一些简单的分子动力学过程,可能 100 ns 的模拟时长就足够,但对于复杂的生物过程或材料性能变化研究,可能需要更长的模拟时间,甚至达到微秒级或更长。
运行模拟
完成系统初始化和模拟参数设置后,就可以使用专门的 MD 模拟软件求解运动方程。这些软件根据输入的分子结构、力场参数和模拟条件,通过数值计算方法求解牛顿运动方程,计算每个原子在不同时刻的位置和速度,并将这些信息记录在轨迹文件中。
轨迹文件如同分子运动的 “日记”,详细记录了分子在模拟过程中的动态变化,为后续的分析提供了丰富的数据。
后处理分析
后处理分析是从模拟轨迹数据中提取有价值信息的关键步骤。在结构分析方面,径向分布函数(RDF)能够反映分子周围原子的分布情况,通过计算 RDF,可以了解分子间的距离分布、局部结构特征等;氢键网络分析则有助于揭示生物分子中氢键的形成和断裂过程,对于理解蛋白质的折叠、核酸的碱基配对等生物过程具有重要意义。
动力学性质分析主要关注分子的扩散系数和均方位移(MSD)。扩散系数描述了分子在体系中的扩散能力,通过计算扩散系数,可以研究分子在溶液中的扩散行为、分子间的相互作用对扩散的影响等;均方位移则反映了分子随时间的位移变化情况,是衡量分子热运动剧烈程度的重要指标。
自由能计算是 MD 模拟中的一项重要分析内容,常用的方法包括伞状采样或 Metadynamics。自由能计算能够帮助研究人员了解分子在不同构象之间转换的能量障碍,对于研究化学反应的机理、蛋白质的折叠路径等具有重要意义。
常用软件对比

三种常见软件的使用方法
GROMACS
GROMACS 是一款在生物分子模拟领域广泛应用的软件,其使用流程具有一定的规范性和系统性。首先,需要准备拓扑文件(.top)和结构文件(.gro)。拓扑文件定义了分子的连接方式、原子类型等信息,就像分子的 “建筑蓝图”;结构文件则记录了分子中每个原子的初始位置。
然后,使用 grompp 工具生成模拟输入文件,该工具会根据拓扑文件、结构文件和用户设置的模拟参数,整合生成包含所有模拟信息的输入文件。接着,运行 mdrun 命令进行模拟,mdrun 会根据输入文件中的信息求解运动方程,生成分子运动轨迹。
最后,通过 gmx rdf 或 gmx msd 等命令对轨迹进行分析,获取如径向分布函数、均方位移等有价值的数据。GROMACS 适用于蛋白质折叠、脂膜动力学、溶液中的小分子行为等研究领域,其高性能并行计算能力和与 VMD 可视化工具的集成,为研究人员提供了便捷高效的模拟和分析平台。
官网:https://www.gromacs.org/
AMBER
AMBER 软件在药物设计和生物分子研究方面具有独特的优势。使用 AMBER 进行模拟时,首先要用 tleap 工具生成拓扑和坐标文件(.prmtop 和.inpcrd)。tleap 能够根据用户指定的分子结构和力场参数,自动生成包含分子拓扑信息和初始原子坐标的文件。
接着,编写输入文件(.mdin),在该文件中详细定义模拟参数,如温度、压力、模拟时长等。然后,运行 sander 或 pmemd 程序执行模拟,这两个程序会根据输入文件和拓扑坐标文件,进行分子动力学计算。
最后,使用 cpptraj 工具对模拟轨迹进行分析,cpptraj 提供了丰富的分析功能,能够帮助研究人员从轨迹数据中提取各种结构和动力学信息。AMBER 适用于药物 – 靶标相互作用、核酸动力学、酶催化机制等研究领域,其支持 QM/MM 混合计算的特点,使得在研究涉及量子力学和经典力学相互作用的生物过程时具有显著优势。
官网:https://ambermd.org/AmberMD.php
LAMMPS
LAMMPS 是一款在材料科学领域备受青睐的软件,特别适合处理超大规模分子系统的模拟。使用 LAMMPS 的第一步是准备数据文件,包括原子坐标和力场参数。原子坐标文件记录了分子中每个原子的位置信息,力场参数文件则定义了原子间的相互作用势能。
然后,编写输入脚本(.in),在脚本中明确指定积分算法、输出设置等模拟参数。积分算法的选择会影响模拟的准确性和效率,输出设置则决定了模拟结果的记录方式和内容。接着,运行 lmp_mpi 命令启动并行计算,利用多核处理器或集群计算资源,加速模拟过程。最后,可以使用内置命令或外部工具(如 OVITO)对模拟结果进行分析。
OVITO 是一款强大的可视化和分析软件,能够将 LAMMPS 的模拟结果以直观的图形方式展示出来,方便研究人员观察分子结构变化和分析物理性质。LAMMPS 适用于金属玻璃结构、聚合物自组装、纳米材料力学性能等研究领域,其对超大规模系统的支持能力,为研究复杂材料体系的微观结构和性能提供了有力工具。
官网:https://www.lammps.org/
总结
分子动力学模拟凭借经典力学模型,为我们揭示了分子尺度下的动态行为奥秘,其应用范围广泛覆盖生物、材料和化学等众多领域。在实际应用中,软件的选择至关重要,需要综合考虑体系规模、精度需求和计算资源等因素。
例如,GROMACS 凭借其高性能并行计算和可视化优势,成为生物大分子模拟的理想选择;LAMMPS 则以其对超大规模系统的支持,在材料模拟领域独树一帜;AMBER 在药物设计中,利用其 QM/MM 混合计算能力,展现出独特的优势。随着科学技术的不断发展,未来分子动力学模拟的发展趋势将聚焦于多尺度建模和机器学习力场的应用。
多尺度建模能够将微观分子行为与宏观材料性能相结合,提供更全面的研究视角;机器学习力场则有望通过对大量数据的学习,构建更准确、高效的分子间相互作用模型,进一步推动分子动力学模拟技术的发展和应用。
以上扩写详细呈现了分子动力学模拟的多方面内容。你对扩写内容的篇幅、某些部分的深度是否满意,或者还有其他修改方向,都能随时告诉我。