

分子动力学模拟(Molecular Dynamics, MD)是一种通过经典力学方法研究原子和分子随时间演化行为的计算模拟技术。
其基本思想是根据牛顿第二定律,利用原子间相互作用的势能函数(力场),计算每个原子的受力,从而推导出其加速度、速度和位置随时间的变化。
通过对大量原子的运动进行逐步积分,可以模拟出整个系统的结构变化、热力学性质、扩散行为、相变过程等。
分子动力学模拟广泛应用于化学、物理、生物学、材料科学等领域,比如模拟蛋白质折叠、研究液体中的扩散行为、预测材料在不同温度或压力下的性能等。
相较于静态的量子化学计算,MD模拟能提供关于系统动态演化的详细信息,更加贴近实验过程。
但需要注意的是,传统分子动力学模拟基于经典力学,难以描述电子结构变化,因此在处理涉及化学反应或激发态过程时,常需结合量子力学方法或使用更复杂的反应力场。
分子动力学模拟的核心原理
分子动力学模拟基于牛顿运动方程,通过数值积分计算原子或分子在势能场中的运动轨迹。其核心方程可表示为:


其中,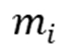 和
和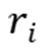 分别为第
分别为第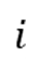 个原子的质量和位置,
个原子的质量和位置,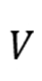 是体系势能函数。
是体系势能函数。
通过时间离散化(时间步长通常为飞秒量级),模拟系统内所有原子在力场作用下的动态演变。
势能函数的选择(如Lennard-Jones势、Coulomb势或量子力学修正的力场)直接决定了模拟的精度与适用范围。
例如,经典力场适用于大体系的长时模拟,而ReaxFF力场则能描述化学键的断裂与形成,适用于反应动力学研究。
关键算法:在分子动力学模拟中,Verlet算法和Leapfrog算法因其出色的数值稳定性和能量守恒性,被广泛用于积分牛顿运动方程。
以Verlet算法为例,其基本思想是利用当前位置和前一时间步的位置,结合当前的合力 F(t)F(t)F(t),来预测下一步的原子坐标,原子位置更新公式为:

这种方法避免了对速度的显式存储,仅依靠位置进行更新,从而有效降低了内存占用,特别适用于大规模原子体系的模拟。
此外,Verlet算法具有较好的时间可逆性和对系统总能量的长期稳定控制,不易积累误差,使其在长时间模拟中表现优异。
因此,它成为GROMACS、LAMMPS等主流MD软件中的默认积分器之一,尤其适合进行大规模并行计算和生物分子模拟。
多尺度应用与可视化案例
力学性能预测与缺陷演化
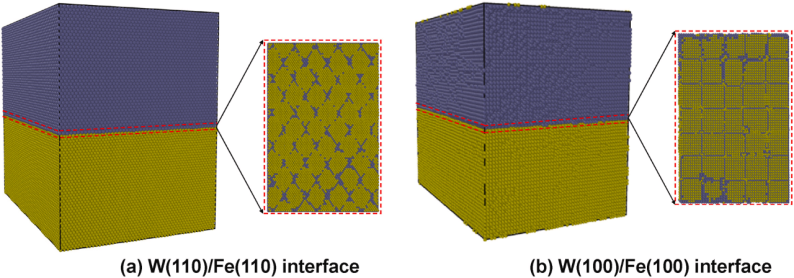
在W/Fe界面的研究中,研究者构建了W(110)/Fe(110)和W(100)/Fe(100)两种半共格界面模型,采用分子动力学模拟的方法,通过LAMMPS软件包和自行开发并修正的Fe-W嵌入原子法(EAM)势,结合ZBL势来准确预测级联效应,对W/Fe半共格界面在辐照下的缺陷演化和力学性能进行了系统研究。
模拟了在辐照过程中纯钨、纯铁以及两种半共格界面的位错环演化和应力–应变曲线,分析了不同界面取向对辐照抗性的影响,为W-Fe系统在聚变环境下的设计提供了指导。
(https://doi.org/10.1016/j.vacuum.2022.111618)
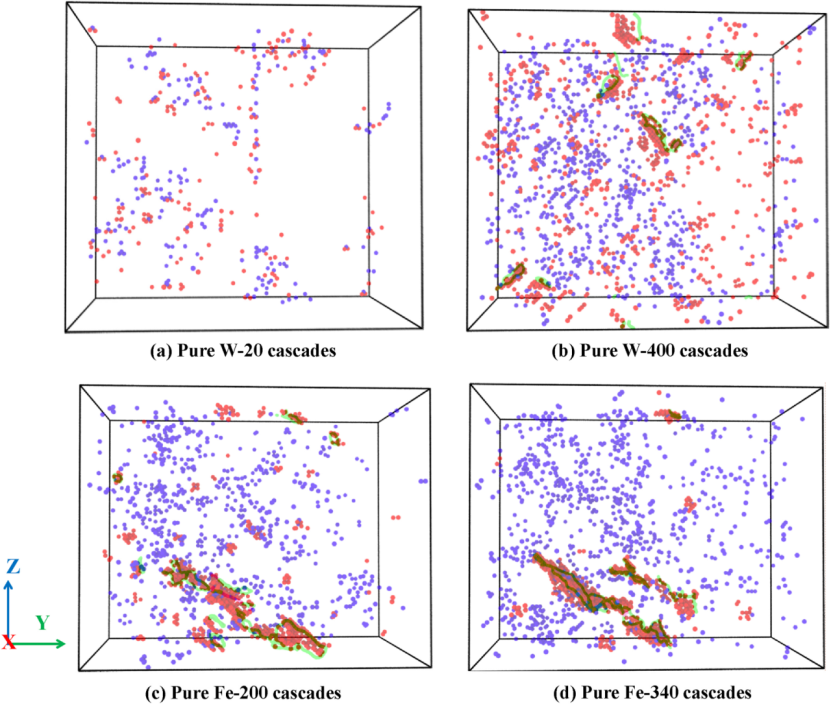
图像解析:下图展示了纯钨和纯铁在不同损伤阶段的点缺陷分布和形成的位错环。
图中蓝色和红色球分别代表空位和间隙原子,绿色和粉色线分别表示在辐照下形成的柏氏矢量为1/2和的位错环。
从图中可以看出,随着辐照次数的增加,纯钨和纯铁中的间隙原子不断积累,最终导致位错环的形核。
在纯铁中几乎只发生位错环,而在纯钨中则只观察到柏氏矢量为1/2的位错环,这与实验观察结果一致。
这表明辐照导致的损伤积累会引发位错环的形成和扩展,进而影响材料的力学性能。
(https://doi.org/10.1016/j.vacuum.2022.111618)
纳米材料设计与性能优化
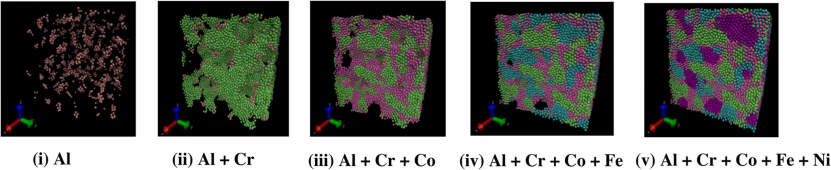
AlCrFeCuNi高熵合金的拉伸模拟展示了弹性–塑性转变的动态过程。
在Al含量为10%时,模拟轨迹显示初始阶段原子规则排列(弹性区),随着应变增加,孪晶(绿色原子簇)和层错(红色原子带)逐渐形成(屈服点),最终导致非均匀塑性变形。
通过对比不同Al含量的应力–应变曲线,发现杨氏模量随Al含量增加呈线性下降,这一结果与X射线衍射测得的晶格常数变化高度吻合。
(Sci Rep 6, 31028 (2016). https://doi.org/10.1038/srep31028)
涂层与表面特性研究
在材料科学领域,MD模拟通过追踪原子位置与势能变化,为材料设计提供理论指导。
例如,碳化硅(SiC)作为高温半导体材料,其晶体结构的热稳定性直接影响器件性能。研究团队通过全原子MD模拟,比较了Tersoff、Vashishta等势函数在SiC熔融–凝固过程中的预测精度。
模拟结果显示,Tersoff势能准确描述碳硅键的断裂阈值(约2500K),而Vashishta势在界面扩散系数计算中误差低于5%。
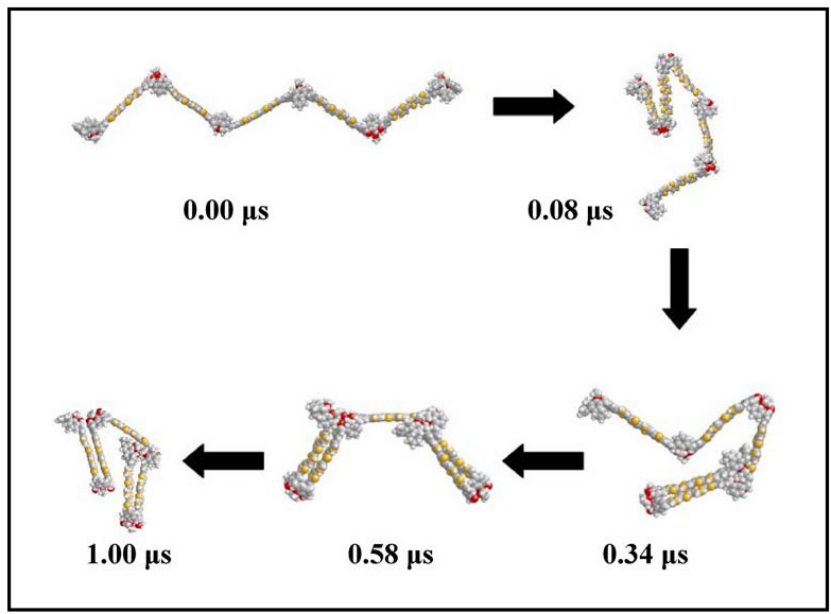
下图展示了纳米执行器在1微秒收缩过程中的结构演变:初始阶段(0.00μs)原子排列松散,随着时间推移,局部应力促使键角调整,至1.00μs时形成致密网状结构。
这种动态可视化揭示了材料在机械载荷下的自组织行为,为设计耐高温涂层提供了原子级依据。
聚合物摩擦学研究中,MD模拟揭示了石墨烯增强复合材料的减摩机制。
下图展示单晶镍纳米切削过程:刀具前角(绿色标注)与工件原子(蓝色)接触时,位错沿密排面(111)扩展,形成剪切带。
模拟发现,石墨烯片层插入聚四氟乙烯基体后,界面滑移阻力提升30%,归因于碳–氟键的电荷再分布(通过Lennard-Jones势与库仑势耦合计算)。
此类研究为开发低磨损复合材料提供了分子设计策略。

(doi: 10.1007/s11431-016-0251-y)
生物医学领域的创新突破
在生物医学领域,MD模拟能够捕捉蛋白质构象变化与药物结合的动态路径。
以细胞色素P450酶为例,X射线晶体学仅提供静态结构,而GaMD(高斯加速分子动力学)模拟显示,底物结合诱导活性口袋的α–螺旋发生约15°偏转,形成疏水通道。
下图通过多帧叠加展示了酶–配体复合物的动态:红色氧原子在初始阶段(左上)随机分布,随时间推移(右上及下方子图)聚集于结合位点,形成氢键网络。
这一过程与实验测得的结合自由能(ΔG=-9.8 kcal/mol)高度吻合,证实了MD在预测药物亲和力中的可靠性。

化学反应机理与工业催化
MD模拟结合增强采样技术,能够解析复杂反应网络的动态细节。
例如,沸石催化甲醇制烯烃(MTO)过程中,研究者采用ReaxFF力场模拟了甲基苯生成路径。
下图展示了自由基(红色球体)从FTC态(富电子)向ETC态(缺电子)的转化:初始阶段(上半部分),自由基在范德华力作用下接近活性位点;进入反应器(下半部分)后,绿色催化剂原子诱导C-H键断裂,生成中间体(蓝色球体)。
模拟结果与同位素标记实验的产物分布一致,验证了“碳池”机理的合理性。
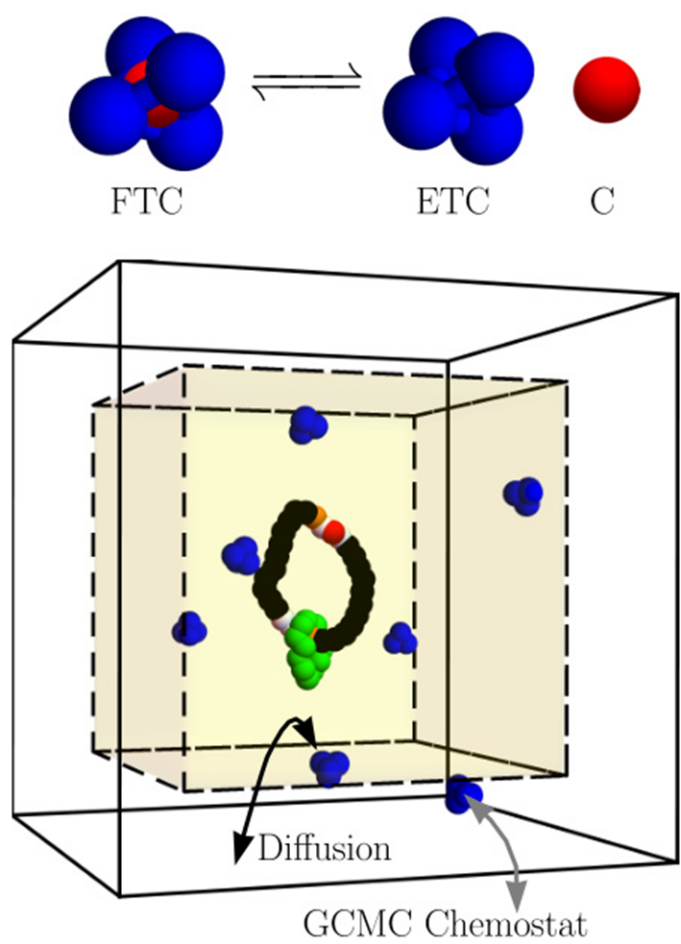
环境污染物迁移与界面行为
环境科学中,MD模拟揭示了大气颗粒物形成与吸附的原子机制。
下图整合了分子模拟(左)与气候模型(右),展示二次有机气溶胶(SOA)的生成:挥发性有机物(VOCs,灰色球体)在液滴表面(蓝色界面)发生缩合反应,形成核心–壳结构(黄色球体)。
模拟发现,表面张力降低(从72 mN/m至45 mN/m)可促进粒径增长速率达20%。
此外,MD模拟还量化了重金属离子在黏土矿物层间的迁移能垒,为土壤修复剂设计提供了理论支持。
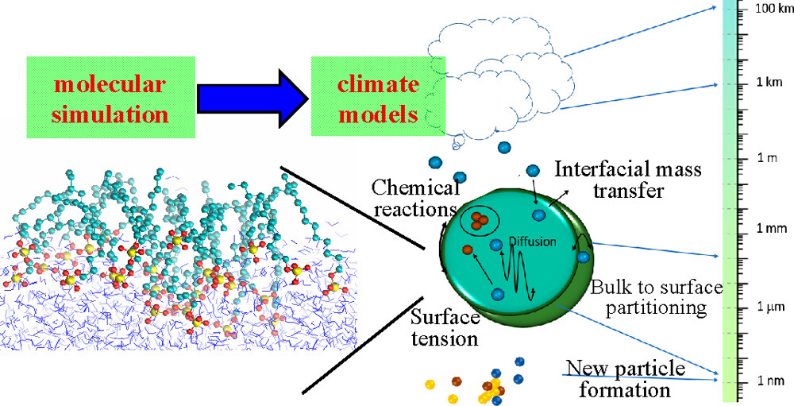
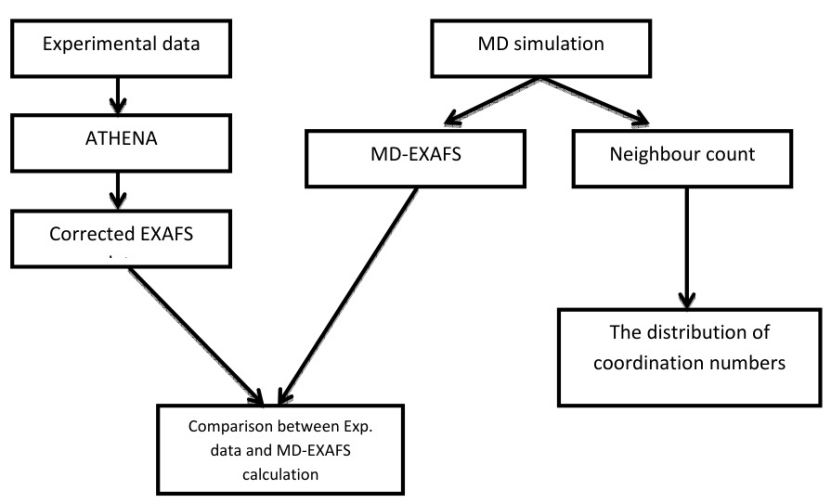
技术演进与跨尺度模拟
MD模拟的实现依赖于高效算法与并行计算架构。
以GROMACS为例,其多线程MPI库将万核计算的效率提升至90%,使微秒级蛋白质折叠模拟成为可能。
CHARMM力场通过极化模型改进,将水分子偶极矩误差从30%降至5%,显著提升生物膜渗透性预测精度。
下展示了软件工具链的整合流程:实验数据(EXAFS)与MD轨迹(配位数分布)的对比验证了模拟可靠性,误差低于0.02 Å。
同时,机器学习势场(如DeePMD)的引入,使模拟精度接近DFT水平,而计算耗时仅增加20%。
挑战与未来方向
尽管MD模拟已在多个领域取得突破,其时间尺度(通常限于微秒级)和力场精度仍是主要瓶颈。
融合量子力学/分子力学(QM/MM)的多尺度方法,以及基于主动学习的自适应力场,有望进一步拓展其应用边界。
例如,在高温合金设计中,结合MD模拟与相场法,可跨越从原子扩散到宏观相变的多个尺度,为材料设计提供全链条指导。
通过上述跨学科案例可见,分子动力学模拟不仅是理论研究的“数字显微镜”,更是工程优化的“虚拟试验场”。
从原子位移的细微变化到宏观性能的定量预测,其能力边界正随着算法革新与算力提升不断扩展。