

总结:过渡金属因其独特的d轨道结构和多变的氧化态,被誉为催化领域的“活性明星”,几乎主导了现代催化剂的设计和应用。
从d带中心理论出发,科学家们揭示了过渡金属催化活性的本质,并通过精准调控其电子结构,实现了高效、可调的催化反应。
本文华算科技结合最新研究进展,系统解析了过渡金属为何能成为高效催化中心,以及d带中心理论在催化剂开发中的应用实例,为设计新一代高性能催化剂提供了理论指导和创新思路。
众多催化材料中,有一类元素堪称“超级明星”,它们就是元素周期表中间区域的过渡金属(如铁、钴、镍、铜、铂、钯、铑等)。为什么它们如此特别,能够成为最高效的催化活性位点呢?这背后隐藏着深刻的电子结构奥秘。
今天,我将带大家一起,从一个核心理论——d带中心理论(d-band center theory)出发,结合最新的前沿研究,揭开这个谜底。
过渡金属的“天赋异禀”:独特的d电子轨道


要理解过渡金属的催化魔力,我们必须先深入到原子层面,看看它们的电子排布。与主族元素不同,过渡金属最外层的电子轨道中,有一个未被完全填满的d轨道。这个“部分填充”的状态,是它们一切特性的根源。
灵活的“手”:这些d轨道上的电子(我们称之为d电子)既不像内层电子那样被原子核牢牢束缚,也不像最外层s电子那样容易失去。它们处于一种非常灵活的状态,可以方便地与外来分子(反应物)形成化学键,又能适时地断开化学键,释放产物。这种“收放自如”的能力,正是催化循环所必需的。
多变的“性格”:过渡金属通常具有多种稳定的氧化态(例如,铁可以是+2价或+3价)。这意味着在催化反应中,它们可以灵活地给出或接收电子,充当电子转移的中转站,从而为复杂的化学转化提供通道。
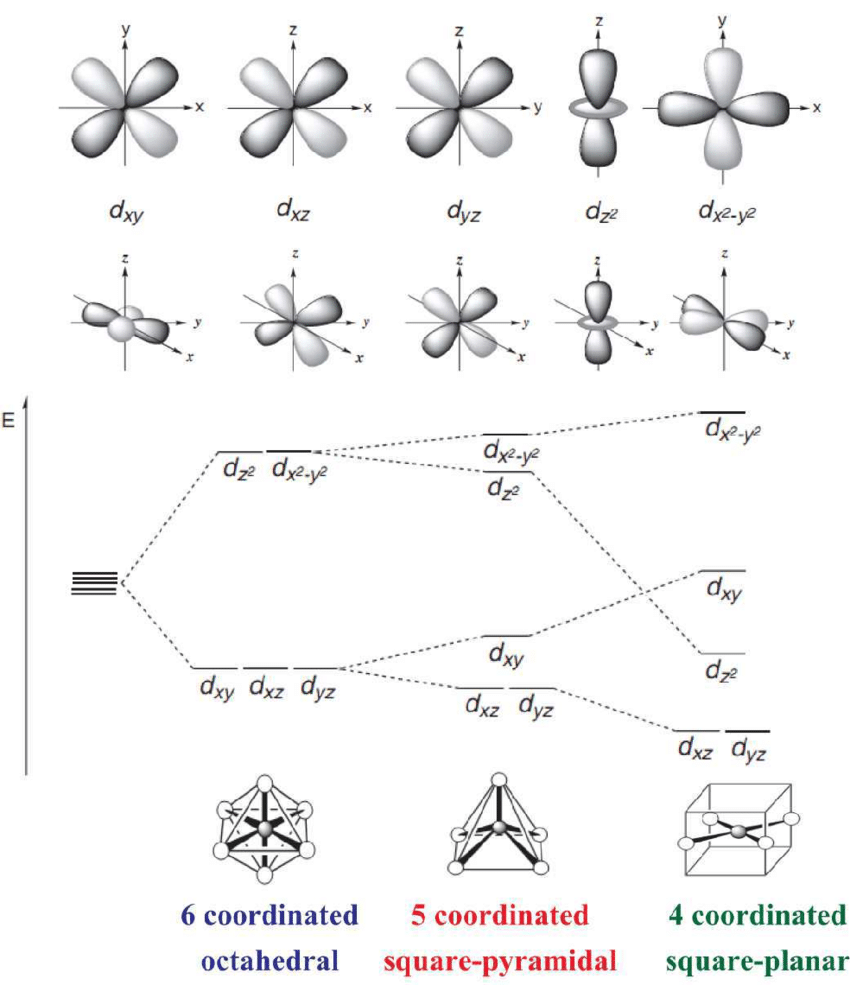
这些独特的电子特性,使得过渡金属表面能够像一个“活性平台”,吸引反应物分子前来“落脚”,并降低它们发生化学反应所需的能量门槛(即活化能)。
但是,不同的过渡金属催化性能千差万别,我们如何量化和预测它们的催化活性呢?这就引出了我们今天的主角——d带中心理论。
d带中心理论:衡量催化活性的“黄金标尺”
d带中心理论最早由Jens Nørskov和B.Hammer等科学家在20世纪90年代系统性地提出和发展,如今已成为催化剂设计的核心指导思想之一。
这个理论的核心思想非常精妙:过渡金属表面d电子的平均能量(即d带中心)决定了其与反应物分子的结合强度,而这个结合强度又直接关系到最终的催化活性。
我们可以用一个简单的比喻来理解:
d带中心的位置:想象d电子的能量是一个“平台”的高度。d带中心能量越高,这个“平台”就越高,表明d电子越活跃,越容易与吸附上来的分子形成强的化学键。
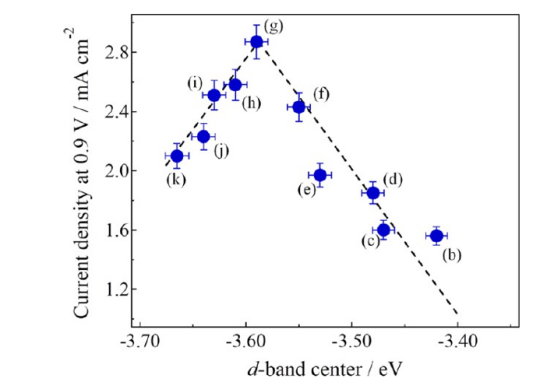
结合强度的“金发姑娘原则”:催化反应的效率遵循一个所谓的“火山型关系”或“萨巴蒂尔原则”(Sabatier principle)。反应物与催化剂表面的结合不能太弱,否则反应物根本无法被有效活化;但也不能太强,否则产物会“赖着不走”,占据活性位点,导致催化剂“中毒”失效。最佳的催化剂,其结合强度应该“恰到好处”。
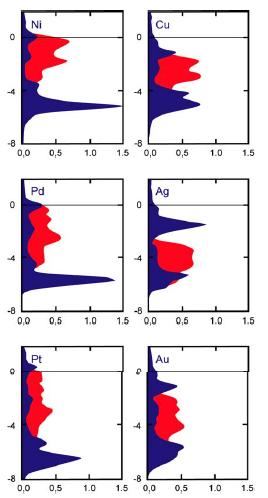
因此,d带中心理论为我们提供了一把“标尺”:通过计算或测量一个过渡金属的d带中心位置,我们就可以预测它与特定反应物的结合强度,并判断其催化活性是否接近“火山”的顶峰。
一个理想的催化剂,其d带中心应该被精确调控到对应火山图顶峰的位置。
从理论到实践:前沿研究中的d带中心理论验证
d带中心理论不仅仅是理论物理学家的“纸上谈兵”,这些年,全球的科研工作者通过大量精密的实验,一次又一次地验证并应用了这一理论来设计新型高效催化剂。
首先,在电解水析氧反应(OER)中有一个典型例子。镍的磷化物Ni2P本身是一种OER催化剂,但活性并不突出。2021年发表在《ACS Catalysis》上的研究以d带中心理论为指导,在Ni2P晶格中引入适量铁元素,合成了Ni-Fe磷化物纳米片。
铁的掺杂令Ni2P的电子结构发生有益改变:实验和理论计算表明,Fe掺入后Ni2P的d带中心适度上移,催化剂对氧中间体的吸附能力明显增强。
但这种提升控制在合理范围内,既不导致吸附过强而难以释放产物,也弥补了纯Ni2P吸附偏弱的不足。
通过这一电子结构调控,该Ni-Fe磷化物纳米片在碱性介质中的析氧活性大幅提高——达到10mA·cm-2电流密度仅需约166mV过电位,明显优于未掺杂的Ni2P催化剂。
这证明了d带中心理论在非贵金属催化剂设计中的指导价值:适度上调过渡金属d带位置,可以平衡催化过程中对中间体的“抓”和“放”,使反应效率接近最佳。
DOI: 10.1021/acscatal.1c01868
在氧还原反应(ORR)领域,d带中心调控同样发挥关键作用。2022年发表于《Small》杂志的一项研究构筑了一种PtCo-PtSn异质结构纳米催化剂:由PtCo和PtSn两种合金相紧密结合,形成富含界面的颗粒。
界面处的强相互作用带来了特殊的电子效应。X射线光电子能谱(XPS)结果显示,与单一PtCo或PtSn相比,这种PtCo-PtSn异质结构中Pt的4f峰显著负移(结合能降低),而Co2p和Sn3d峰正移,表明Pt从周围元素获得电子(发生电子转移)。
Pt d轨道填充度提高,d带中心相对纯Pt/C发生下移——理论计算结果也印证了这一点。适度的d带下移削弱了铂表面对氧中间体的过强吸附,从而更接近ORR反应“火山”曲线的最佳区域。
电化学测试印证了这一点:PtCo-PtSn/C的ORR半波电位约0.93V(高于Pt/C的≈0.90V),在0.90V时的质量活性约1158mA·mg-1(Pt),接近Pt/C的10倍。这些结果表明,通过合金化和界面工程下调d带中心,可显著提升铂基催化剂的ORR性能。
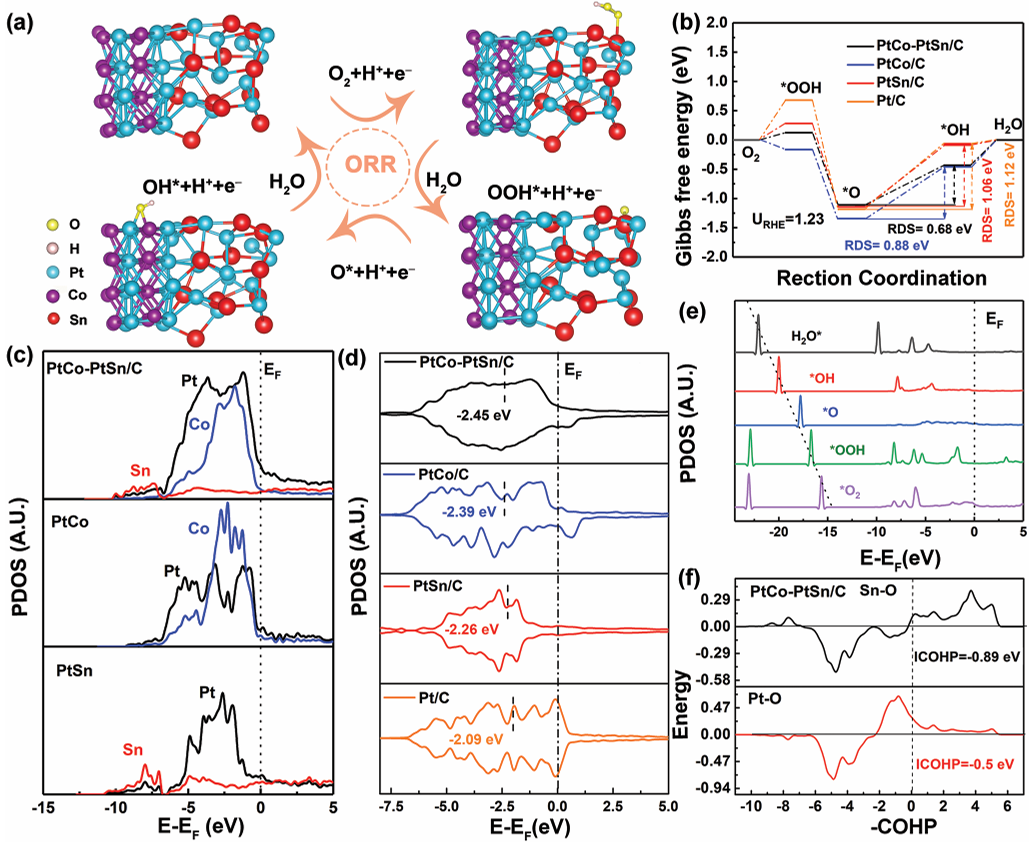
DOI: 10.1002/smll.202106773
最近的研究进一步验证了d带中心调控的巨大潜力。2023年底发表在《Nature Communications》上的一项工作中,研究人员制备了一种特殊的核壳结构,以纯粹的配体效应精确调节铂的d带中心:他们采用与Pt晶格完全匹配的Pd3Ru合金为核,在其上沉积1~2层原子厚的Pt壳。
由于核壳晶格匹配,没有应力影响,这种设计能将性能提升直接归功于电子结构变化。结果显示,Pd3Ru核向Pt壳提供电子,使Pt 5d轨道电子密度增加,Pt的d带中心相对于纯Pt显著下移(约0.2eV)。
这一下移幅度与理论预测的ORR活性最佳值吻合。果然,该核壳催化剂表现出卓越的ORR活性:在碱性介质中0.90V处每毫克Pt可产生约十安培的电流,是商业Pt/C的数十倍,在酸性介质中同样远超Pt/C,并且在长期循环后仍能保持大部分活性。
该研究清晰证明:通过异质合金提供电子,使铂的d带中心达到理想位置,可以令铂基催化剂的ORR性能成倍提高。
DOI: 10.1038/s41467-024-50332-x