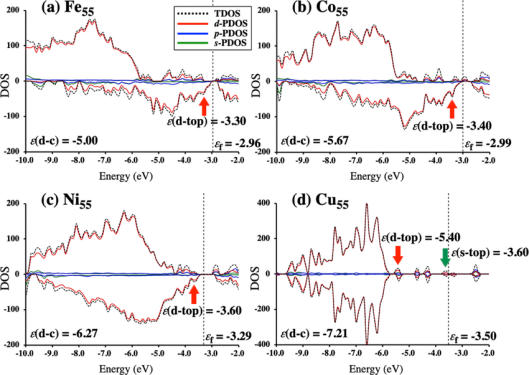
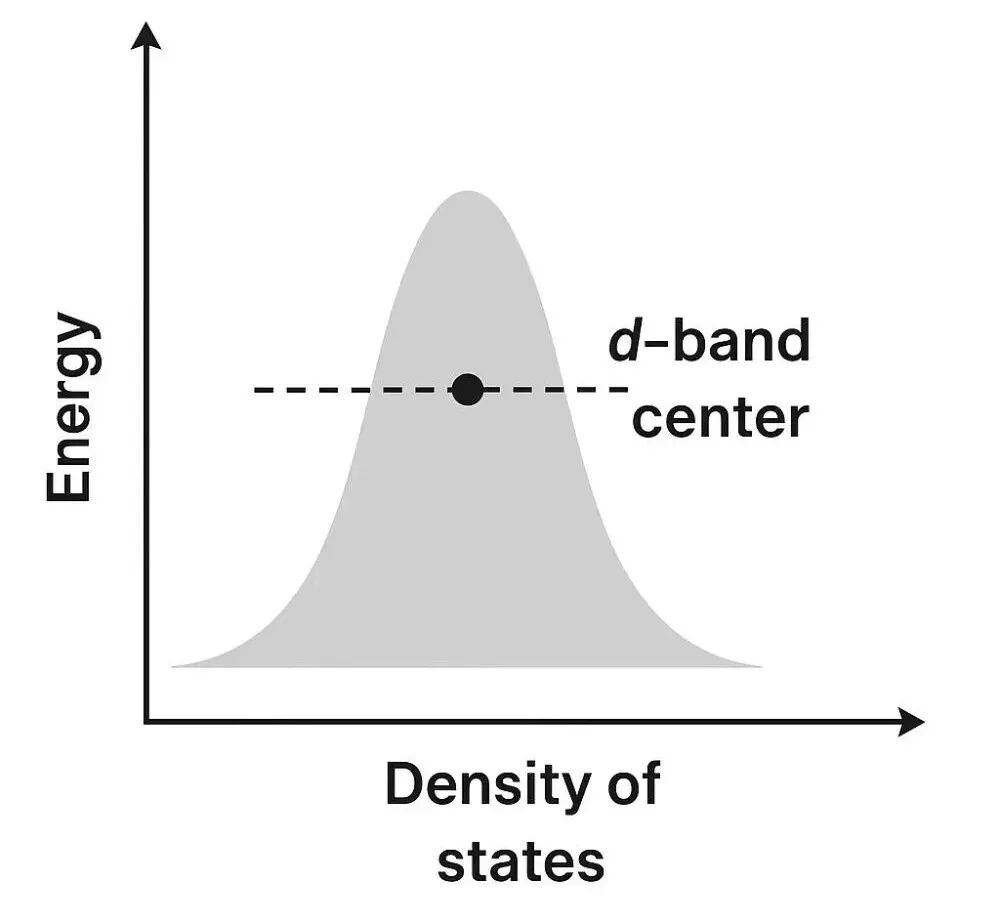
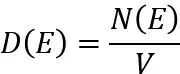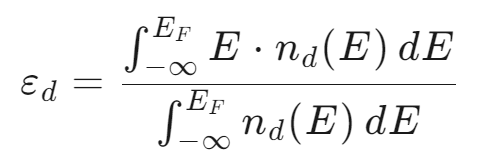d带中心是描述过渡金属原子中d电子平均能量水平的一个物理量。简单来说,d带中心的值越高,通常意味着该金属表面对外来分子的吸附能力越强,化学反应活性也越高。
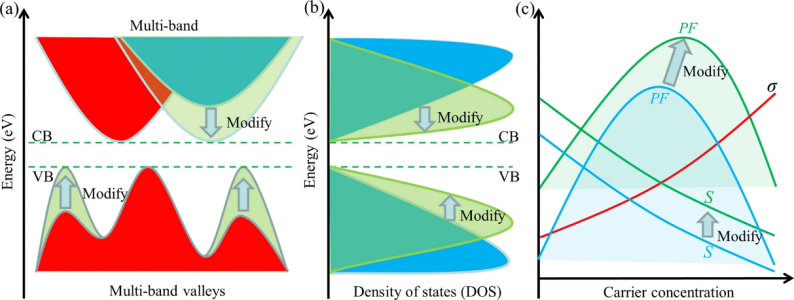
在固体中,单个原子的离散能级会相互重叠,形成连续的能量范围,称为“能带”。对于金属,这些能带部分被电子占据。
而过渡金属的d电子壳层未填满,其许多独特性质如催化、磁性都源于d电子,由d轨道形成的能带就叫d能带,它的能量宽度和位置决定了金属的电子性质。
DOI:10.1038/s41524-020-00476-3
态密度(Density of States, DOS)是固体物理学和凝聚态物理学中的核心概念,用于描述在单位能量范围内电子或量子态的分布情况。其定义为单位频率或单位能量间隔内可被占据的状态数。
具体来说,态密度反映了电子在特定能量范围内的分布密度,是理解材料电子结构、光学性质、电导率等特性的重要工具。
其中,N(E)表示从能量E到E+dE的量子态总数,V是系统的体积。
DOI:10.1021/acsomega.0c05838
d带中心 (d-band center, εd) 在数值上定义为金属表面原子d能带的态密度(DOS)的一阶矩。可以简单理解为:把d能带中所有电子态的能量进行加权平均后得到的位置。
其中:E:能量;nd(E):d轨道的态密度;EF:费米能级。
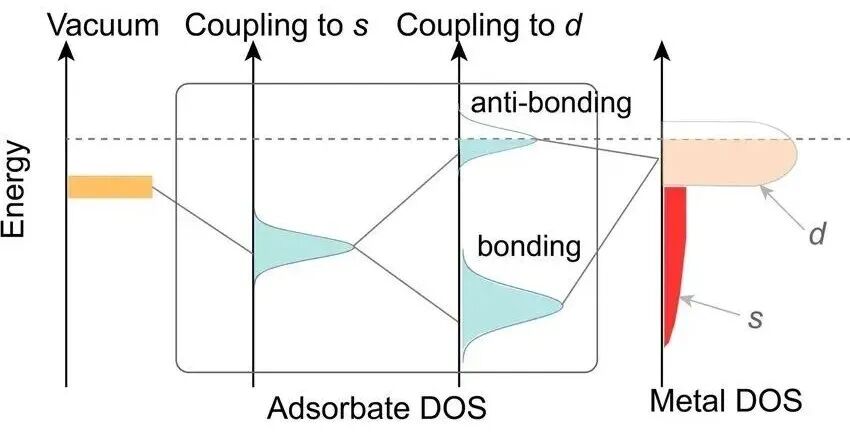
d带中心的重要性在于它直接关联了金属的表面活性和催化性能。一个分子吸附到金属表面时,分子的轨道(通常是σ轨道或π轨道)会与金属原子的轨道发生相互作用,形成两类新轨道:
成键轨道通常会被电子填满,对吸附键的贡献是稳定的,而反键轨道是否被电子占据,则决定了吸附的强弱。
DOI:10.1002/chem.202401718
如果金属的d带中心较高,也就是更靠近费米能级,这意味着金属的d电子本身能量就较高。这会导致反键轨道的能量也被推高。
如果反键轨道被推高到超过了费米能级,那么它就会被排空,没有电子占据。一个被排空的反键轨道意味着整个吸附体系的能量更低,更稳定。因此,金属-分子之间的键会更强。
相反,如果d带中心较低,远离费米能级,反键轨道的能量也较低,容易被电子占据,从而削弱吸附键,使吸附变弱。
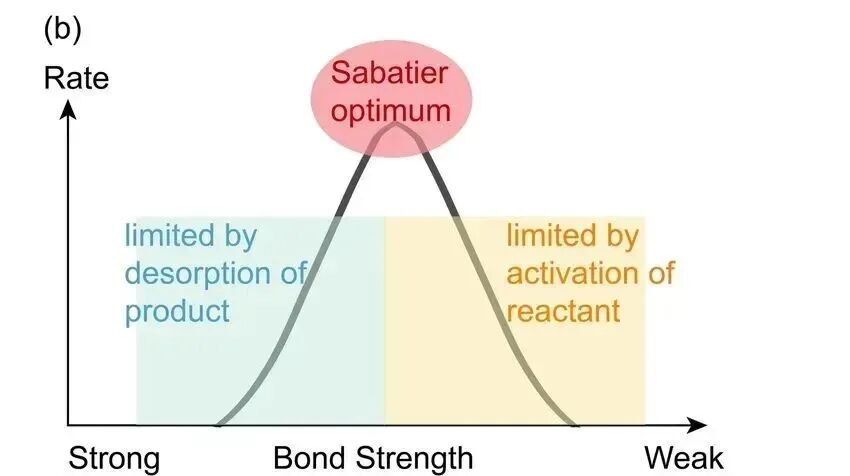
因此,d带中心位置越高,通常活性更高,但过强会使分子无法脱附从而导致催化剂中毒;d带中心位置较低,吸附更弱,活性也会处于较低水平。这就是著名的 Sabatier原理 的电子层面诠释:最佳的催化剂与反应物的吸附既不能太强,也不能太弱。
DOI:10.1002/chem.202401718
合金化通过引入第二种或多种金属元素(如Cu、Zn、Cr)合金化到特定结构,在合金化过程中,不同金属原子之间的相互作用会导致电子的重新分布,进而改变d带中心的位置,提升催化性能。
Pt基催化剂虽活性高但成本昂贵,通过引入Co、Fe等元素形成PtM合金,可通过电子效应降低εd,使H*吸附能接近火山曲线峰值。非贵金属如MoS₂通过边缘位点的d带中心调控,实现了与贵金属可比的HER活性。
DOI:10.1021/acsami.3c00453
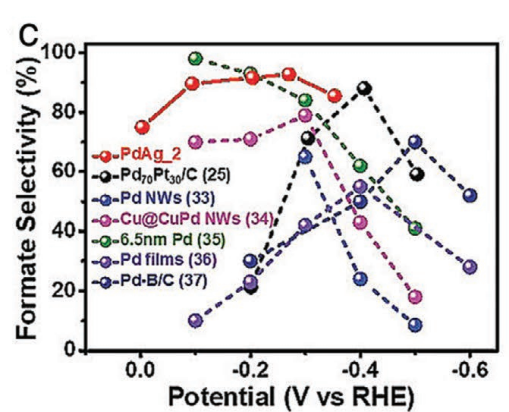
如下图,将Ag与Pd合金化制备的介孔PdAg纳米球,使Pd的d带中心下移,从而提高了对CO中间体的吸附能力,增强了CO2RR的性能。
DOI:10.1002/adma.202000992
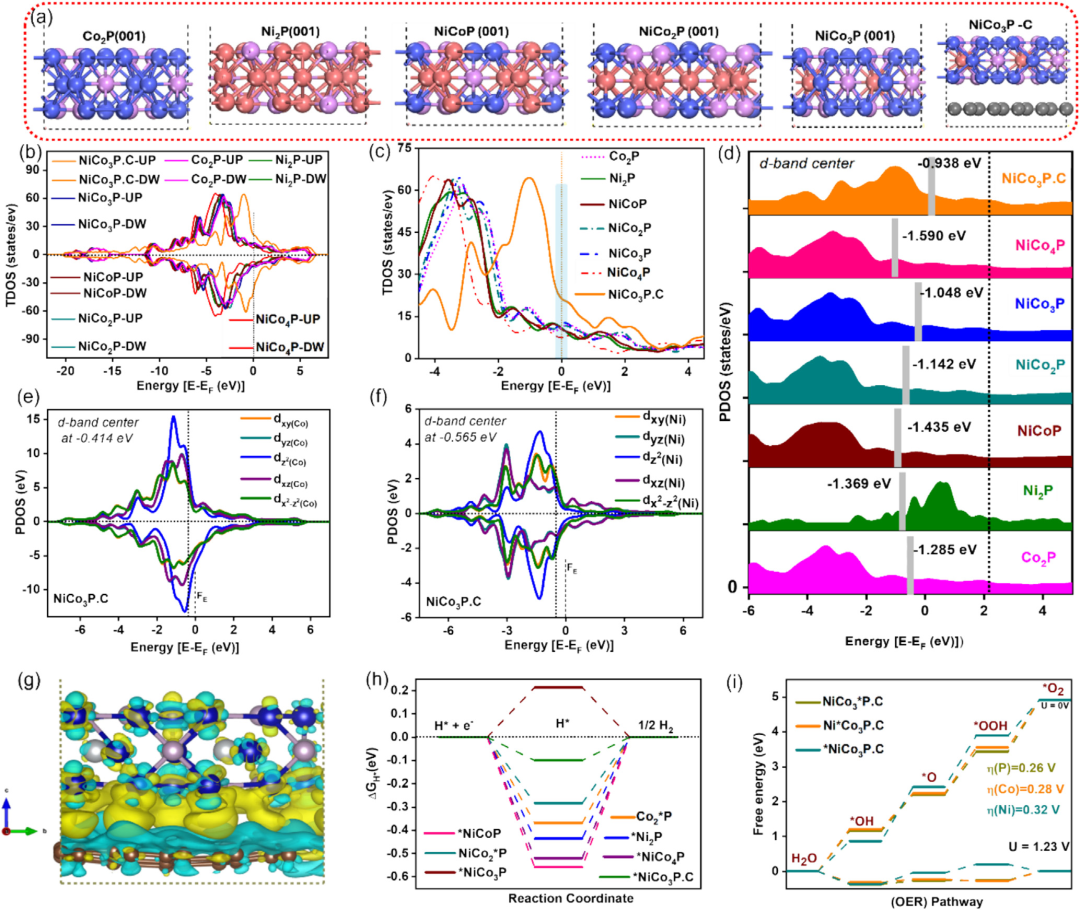
下图中,镍钴磷(NiCo₃P)与还原石墨烯氧化物(RGO)结合后发生了d带中心的上移,催化剂的d轨道与反应物轨道之间的重叠增强,这一变化显著提高了电荷的转移效率。
DOI:10.1016/j.jechem.2024.11.035
拉伸或压缩晶格会改变d轨道重叠程度,从而影响d带中心;
拉伸 → d带变窄 → d带中心上移 → 吸附增强;
压缩 → d带变宽 → d带中心下移 → 吸附减弱。
DOI:10.1021/acs.nanolett.8b04921
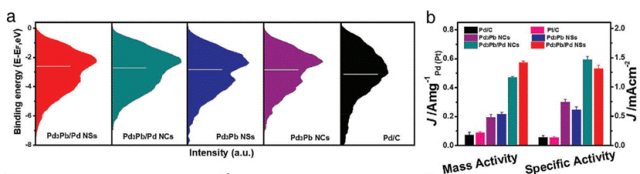
上图中Pd3Pb/Pd核壳纳米片由于存在沿[001]方向的均匀拉伸应变,导致d带中心上移,从而优化了对氧中间体的吸附强度,提高了ORR性能。
表面缺陷、空位、配体修饰等也能改变局部电子结构;如氧空位、氮空位等可提升d带中心,增强吸附。
DOI:10.1002/adma.201904346
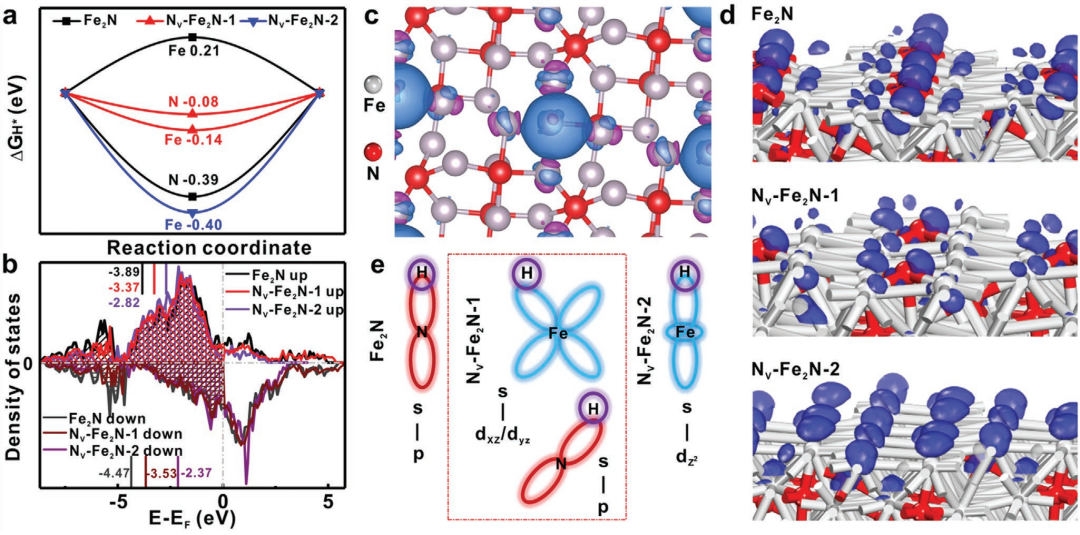
在 Fe₂N 晶格中通过还原去除氮原子生成氮空位,DFT 计算显示,氮空位导致 Fe 的 d-PDOS 峰向费米能级附近移动,价带中心从原始 Fe₂N 的 – 3.89/-4.47 eV(自旋上 / 下)上移至 Nv-Fe₂N-1的 – 3.37/-3.53 eV。
空位的形成会导致电催化剂的电子结构发生变化,使得d带中心位置发生移动。这种电子结构的改变能够影响电催化剂对反应中间体的吸附能力,进而优化电催化性能。
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!