

说明:本文华算科技将系统阐述d带中心的定义、其作为关键描述符的原因,以及该理论如何指导实际的催化剂设计。
什么是d带中心
定义与物理意义
d带中心是催化和表面科学领域,尤其是研究过渡金属催化剂时,一个至关重要的电子结构参数。
从物理上看,d带中心描述的是过渡金属表面原子d轨道电子态的平均能量,其位置通常是相对于费米能级来定义的。
费米能级是电子填充的最高能级,因此d带中心的位置直接反映了金属价电子的能量状态和参与化学键合的“意愿”。
d带中心可以通过第一性原理计算(如密度泛函理论,DFT)获得,其数学表达式为:
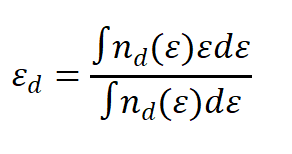

其中,nd(ε)是投影到金属原子d轨道上的电子态密度,ε是能量。
d带中心越高电子越容易填充成键轨道,吸附越强。d带中心越低电子越容易填充反键轨道,吸附越弱。
DOI:10.1021/acsami.3c00453
d带中心的决定因素
金属的d带中心位置受多种因素影响,这为我们调控其催化性能提供了途径。主要影响因素包括:
元素种类:在元素周期表中,从左到右,随着d轨道填充数的增加,d带中心通常会系统性地降低。
应力/应变:当对金属表面施加拉伸应力时,原子间距增大,d轨道交叠减小,导致d能带变窄,d带中心相对于费米能级上移;施加压缩应力则效果相反。
配位环境:表面原子的配位数越低(如台阶、边角、缺陷处的原子),其d带中心通常越高,反应活性也越强。
合金化/掺杂:将两种或多种金属形成合金,或在金属中掺杂其他元素,可以通过电子转移和应变效应显著改变d带中心的位置。
d带中心为何是关键描述符
d带中心之所以被公认为催化领域,特别是多相催化和电催化中的关键描述符,是因为它深刻地揭示了催化剂与反应物分子之间相互作用的本质,并与最终的催化活性和选择性高度相关。
连接电子结构与吸附能
催化反应通常遵循“吸附-表面反应-脱附”的路径。其中,反应物/中间体在催化剂表面的吸附强度是决定反应能否顺利进行的关键。
d带中心理论的核心在于,它成功地将催化剂的电子结构(d带中心)与化学吸附能关联起来。
当一个分子(如CO, O, H等)吸附到金属表面时,其分子轨道会与金属的d轨道发生杂化,形成成键轨道和反键轨道。
d带中心的位置决定了杂化后反键轨道的能量。当d带中心能量升高时,反键轨道的能量也随之升高,若其被推到费米能级之上,则该轨道上的电子填充就会减少,从而导致吸附物种与金属表面之间的净成键作用增强,即吸附能增大。
反之,d带中心降低则导致吸附减弱。这种线性或近线性的相关性,使得d带中心成为预测吸附强度的有力工具。
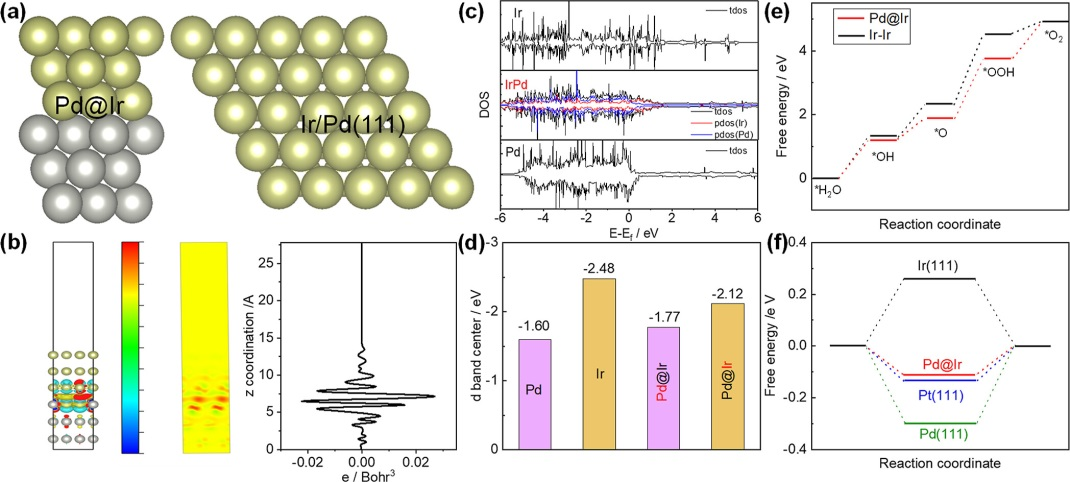
DOI: 10.1039/d1ta02718b
解释催化活性的“火山型”关系
根据萨巴蒂尔原理(Sabatier Principle),一个理想的催化剂对反应中间体的吸附既不能太强,也不能太弱。吸附太弱,反应物难以被有效活化;吸附太强,产物又难以脱附,会“毒化”催化剂表面活性位点。因此,催化活性与吸附能之间常常呈现一种“火山型”关系。
由于d带中心与吸附能密切相关,催化活性自然也与d带中心呈现出类似的火山型依赖关系。这意味着存在一个最优的d带中心位置,能够使催化剂的活性达到峰值。这一发现意义重大,因为它将复杂的催化活性优化问题,简化为寻找并调控一个单一、可计算的电子结构参数——d带中心。
d带中心理论指导催化剂设计
d带中心理论不仅为理解催化现象提供了深刻洞见,更重要的是,它为基于理论计算的理性催化剂设计开辟了道路。
其核心策略是:首先通过理论计算确定目标反应火山曲线的峰顶所对应的最优d带中心值,然后通过各种材料工程手段,合成出具有该最优电子结构的催化剂。
合金化设计: 这是最常用且有效的策略。例如,在ORR催化剂设计中,纯Pt的d带中心略微偏离火山峰顶的强结合端。
通过将Pt与Ni、Co等3d过渡金属合金化,可以利用3d金属的电子效应和应变效应,将Pt的d带中心向更低能量移动,使其更接近火山峰顶,从而获得比纯Pt更高的催化活性。
应变工程: 通过构建核壳结构或将催化剂负载在特定晶格参数的基底上,可以引入拉伸或压缩应变。
例如,在Pd基催化剂上外延生长一层Pt,由于晶格失配,Pt层会受到拉伸应变,使其d带中心上移,从而增强了对某些反应物的吸附和活化能力。
缺陷与界面工程: 在催化剂表面引入原子空位或缺陷,可以改变周围原子的配位环境和电子分布,从而局部调控d带中心。
设计异质结构界面,利用界面处的电荷重新分布,也能有效调节材料的d带中心位置,进而优化催化性能。
单原子催化剂: 在单原子催化剂中,中心金属原子的d带中心受到载体和配位原子的强烈影响。
通过选择不同的载体(如石墨烯、氮化碳、金属氧化物等)和调控配位原子(如N、O、S),可以精确地调节中心原子的d带中心,以适应不同催化反应的需求,如CO₂还原、析氢反应(HER)等。
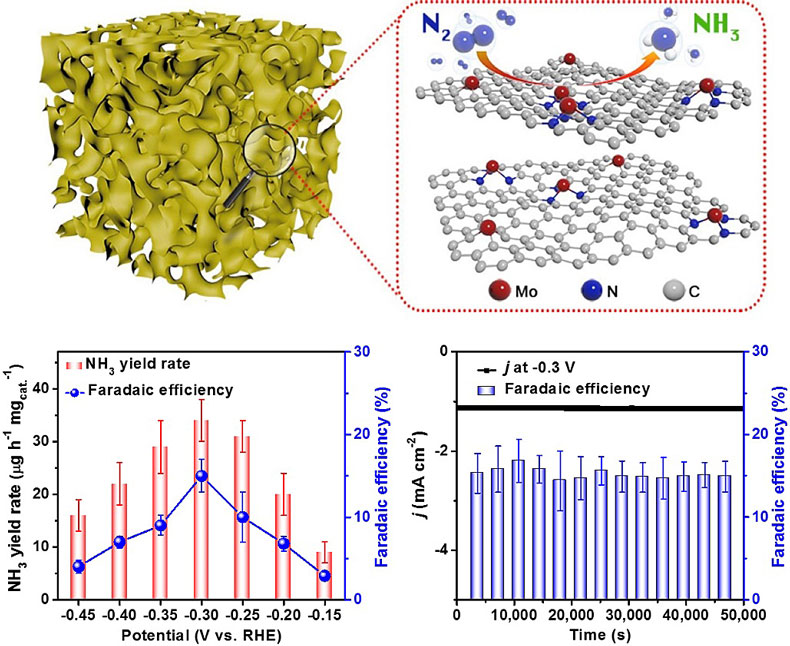
DOI: 10.1002/anie.201811728
结论
d带中心理论是连接微观电子结构与宏观催化性能的基石。它将复杂的催化剂-吸附物相互作用简化为一个可计算、可调控的物理量——d带的平均能量。
通过将d带中心与吸附能和催化活性的火山型关系相结合,该理论为我们从“试错”走向“理性设计”提供了清晰的路线图。
通过合金化、应变工程、缺陷调控等手段精确调节材料的d带中心,已成为当前催化科学领域开发下一代高性能催化剂最重要和最前沿的研究范式之一。
【做计算 找华算】
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???