

说明:本文华算科技系统阐述了材料科学与催化学领域中的核心概念:d带中心。本文将从其基本定义、计算方法及其在催化活性预测等领域的广泛应用三个层面展开,旨在为读者提供一个全面且深入的理解。
什么是d带中心?
d带中心(d-band center)是一个在多相催化、表面科学和电化学等领域中,用于描述过渡金属电子结构特征的关键理论参数。
它并非一个直接的物理可观测量,而是一个通过理论计算得出的描述符(descriptor),其核心技术含义在于量化了过渡金属原子d电子能态的平均能量位置。
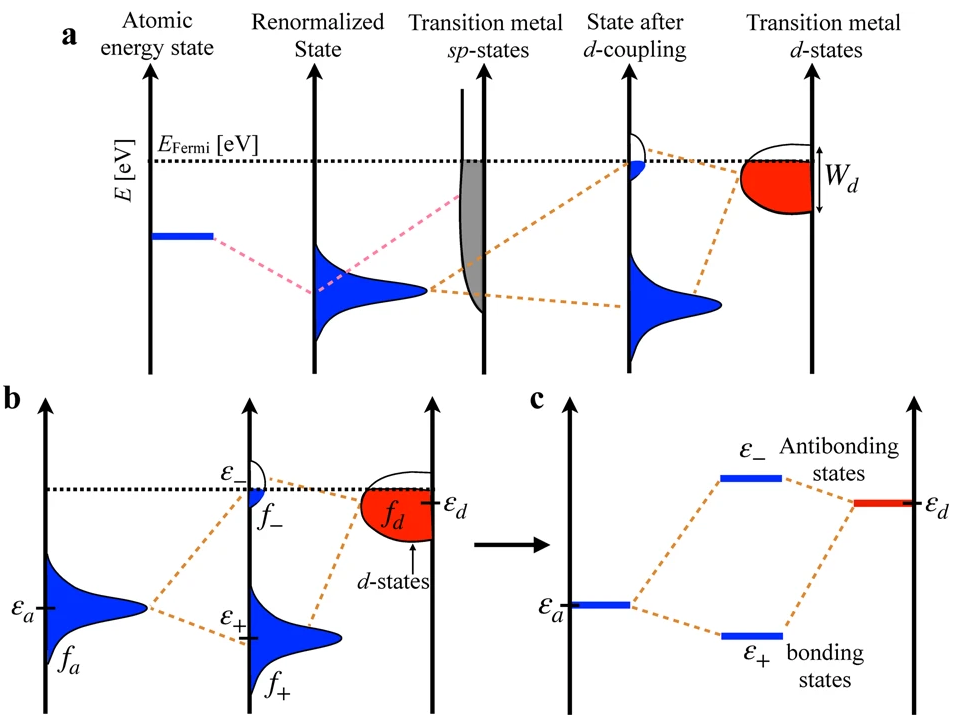

DOI:10.1038/s41524-022-00846-z
根据定义,d带中心是金属d轨道的投影态密度(Projected Density of States, PDOS)相对于费米能级(Fermi Level,EF)的能量加权平均值。简而言之,它指明了d电子态能量分布的“重心”所在。
费米能级代表了电子填充的最高能级,因此d带中心的位置直接反映了d电子态的填充情况以及它们在化学成键中的活跃程度。
d带中心模型的理论基础,尤其是在催化领域的应用,主要由Hammer和Nørskov等人发展和完善。该模型指出,过渡金属表面与吸附物(如CO、O、H等)之间的相互作用强度,与该金属的d带中心位置密切相关。具体而言:
d带中心与成键强度的关系:当吸附物的分子轨道与金属表面的d轨道发生杂化时,会形成成键轨道和反键轨道。d带中心的位置决定了杂化后反键轨道的能量。
一个更靠近费米能级(即能量更高)的d带中心,通常意味着d电子态的能量更高、填充更少,这使得杂化后形成的反键轨道能量也更高,且更多地处于费米能级之上(即未被电子填充)。未被填充的反键态越多,意味着吸附物与金属表面之间形成的化学键越强。
物理意义:因此,d带中心的位置成为了衡量金属表面化学吸附能力的一个有效指标。d带中心越高(越接近费米能级),金属表面与吸附物的作用通常越强;反之,d带中心越低(离费米能级越远),表明d电子态更加稳定、能量更低,与吸附物的作用通常越弱。
这一简单的关联性,使得d带中心成为连接微观电子结构与宏观催化性能的桥梁。
怎么计算d带中心?
d带中心的计算严格依赖于第一性原理(first-principles)的电子结构计算,其中最常用的方法是密度泛函理论(Density Functional Theory, DFT)。计算过程通常包含以下几个步骤:
构建模型与DFT计算:首先,需要构建所研究材料的原子结构模型,例如金属晶体的表面板层(slab)模型。
然后,使用DFT软件(如VASP、Quantum ESPRESSO等)对该模型进行几何优化和静态自洽计算,从而获得体系稳定状态下的电子结构信息,包括总态密度(DOS)和能带结构。
投影态密度(PDOS)的获取:d带中心关注的是特定原子的d轨道电子态。因此,需要从总态密度中分解出目标原子的d轨道的贡献,即获得d轨道的投影态密度(PDOS或LPDOS)。PDOS曲线描述了在不同能量处,d电子态的分布情况。
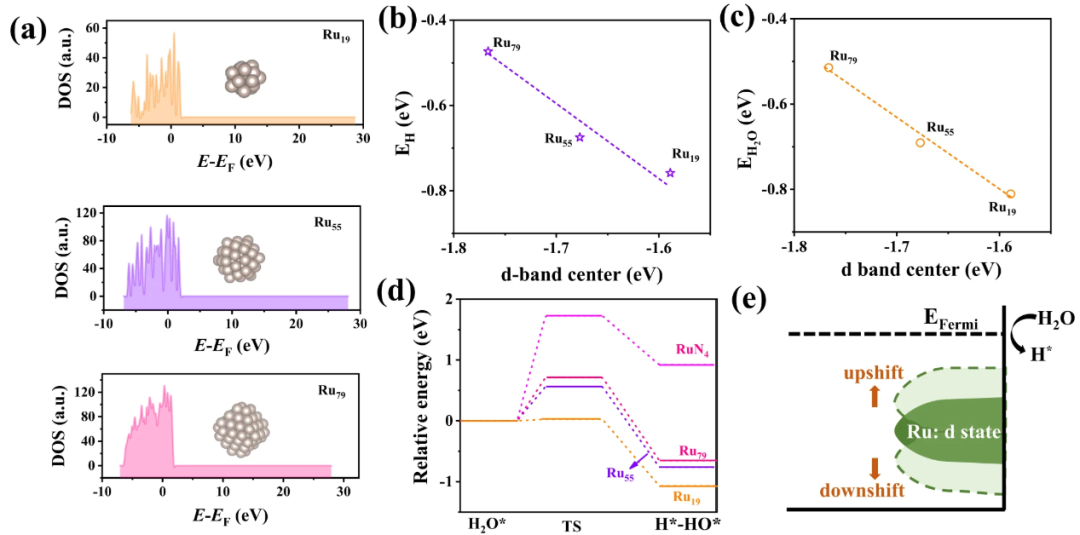
DOI:10.1038/s41467-022-31660-2
积分计算:获得了d轨道的PDOS后,d带中心的值可以通过以下积分公式计算得出:
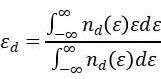
在这个公式中,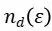 是能量为
是能量为![]() 处的d态投影态密度,分母
处的d态投影态密度,分母![]() 是对所有d电子态的总数进行归一化。整个公式的物理意义就是计算d电子态密度的能量加权平均值。能量
是对所有d电子态的总数进行归一化。整个公式的物理意义就是计算d电子态密度的能量加权平均值。能量![]() 通常以费米能级为参考点(即EF=0eV)。
通常以费米能级为参考点(即EF=0eV)。
通过这个计算,复杂的PDOS曲线被简化为一个单一的数值量,极大地便利了不同材料电子结构之间的比较,并使其能够与催化活性等宏观性质建立起明确的关联。
值得注意的是,由于不同计算方法对角动量投影的定义可能存在细微差异,d带中心的绝对值可能略有不同,但其相对趋势在一致的计算参数下是可靠的 (from results)。
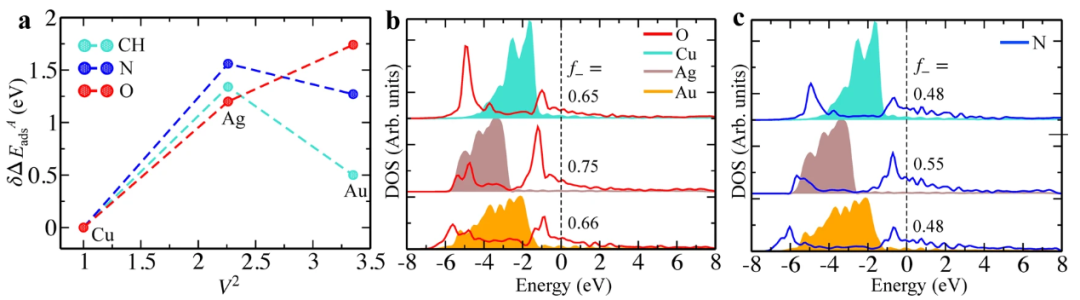
DOI:10.1038/s41524-022-00846-z
d带中心的应用?
催化活性预测与火山图(Volcano Plots):d带中心最成功的应用在于预测和解释过渡金属的催化活性趋势。根据萨巴蒂尔原理(Sabatier Principle),一个理想的催化剂与反应中间体的结合既不能太强也不能太弱。
d带中心恰好可以量化这种结合强度。将一系列不同过渡金属的催化活性(如反应速率)与其d带中心(或与之强相关的吸附能)作图,常常会得到一个“火山形”曲线。
位于火山顶峰的金属,其d带中心处于一个最优位置,对应着中等的吸附强度和最高的催化活性。这一工具使得研究人员能够快速评估和预测新材料的催化潜力。
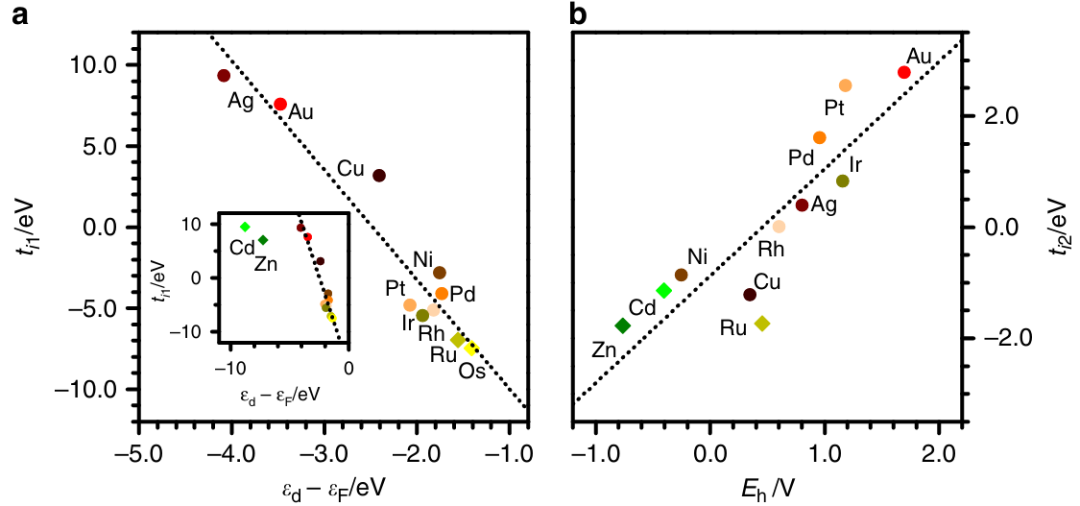
DOI:10.1038/s41467-019-12709-1
合金催化剂的设计:通过将不同金属进行合金化,可以有效调控材料的d带中心位置。
例如,将d带中心较低的金属(如Au、Ag)与d带中心较高的金属(如Pt、Pd)合金化,可以精细调节合金表面的d带中心,使其更接近火山图的顶峰,从而获得比纯金属更高的催化性能。
这为设计高性能、低成本的双金属或多金属催化剂提供了强有力的理论指导。此外,应力/应变工程(strain engineering)也是一种调控d带中心的有效手段。
理解表面反应机理:d带中心不仅能预测活性,还能帮助理解反应机理。通过分析反应路径中各个中间体在不同d带中心表面的吸附能变化,可以判断反应的速控步骤以及表面成分对反应选择性的影响。
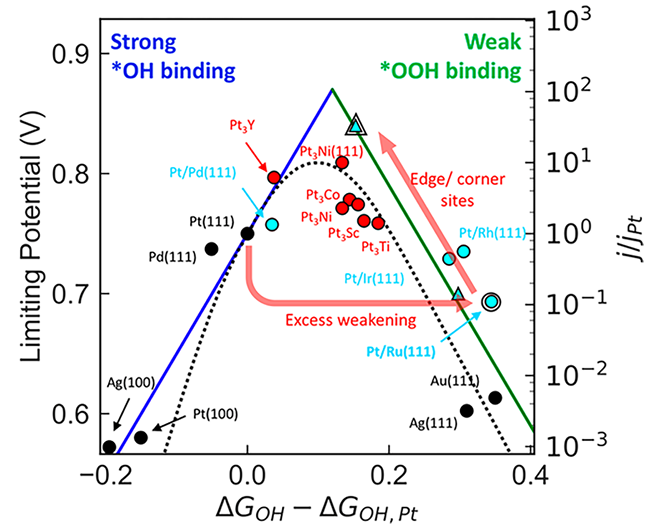
DOI:10.1021/acs.chemrev.7b00488
局限性与发展:尽管d带中心模型取得了巨大成功,但它也存在一定的局限性。例如,该模型在描述某些复杂的反应体系(如OH在Pt/Pd合金上的吸附)时可能会出现例外。
对于单原子催化剂,由于其独特的配位环境和电荷状态,d带中心的适用性仍在深入探讨中。此外,对于具有磁性的过渡金属表面,传统的d带中心模型需要进行扩展,以考虑自旋极化效应对吸附能的影响。
小结
d带中心是描述过渡金属电子结构的关键参数,通过计算d态密度的能量加权平均值得出。它作为催化活性的重要描述符,将微观电子结构与宏观催化性能联系起来,在催化剂的理论设计与筛选中发挥着核心作用,但其应用也存在一定局限性。
【做计算 找华算】
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???