本文系统介绍了溶液中溶剂化结构的定义与特征,重点阐述了DFT计算在解析溶剂化体系中的关键作用。
通过静电势、HOMO-LUMO能级、键长键角、溶剂化/去溶剂化能和结合能等性质的计算,可以深入揭示溶质–溶剂相互作用的微观机制。
例如,静电势分析可预测溶剂化壳层的电荷分布,而结合能计算能量化溶剂化结构的稳定性。
这些理论方法为优化电池电解液、催化反应介质等提供了重要依据。
溶液中溶剂化结构的定义与特征
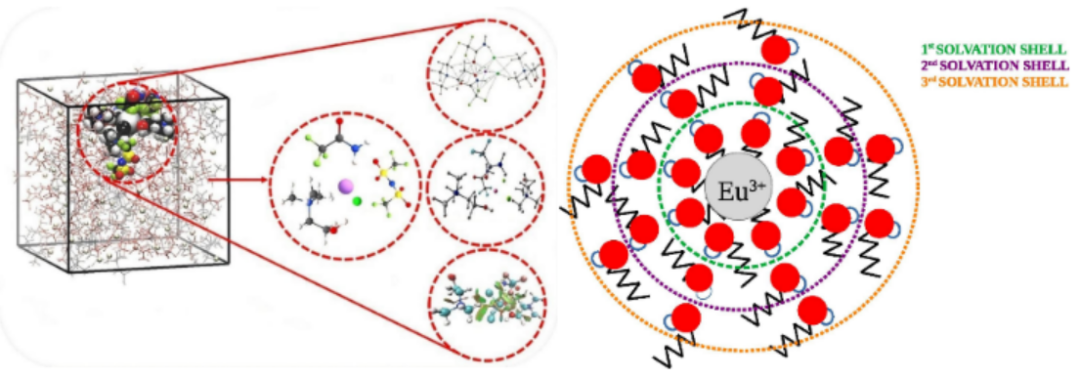
溶剂化结构通常分为第一溶剂化壳层和第二溶剂化壳层。
第一壳层由与溶质直接通过强相互作用(如静电作用、配位键或氢键)结合的溶剂分子构成;第二壳层则包含受溶质影响但未直接配位的部分溶剂分子或离子对。

DFT在溶剂化体系中的可计算性质
1. 静电势(Electrostatic Potential)
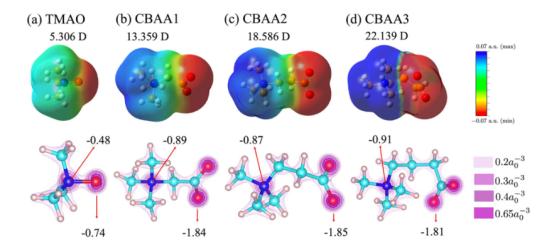
在下图中,静电势计算被用于揭示两性离子化合物(如TMAO和CBAs)的溶剂化结构特性及其抗生物污染机制。
通过分析两性离子的静电势表面(EPS)和偶极矩(TMAO:5.306 D;CBAA3:22.139 D),研究者发现TMAO因缺少疏水间隔基(−(CH₂)ₙ−),其正负电荷中心重叠导致偶极矩最小、静电势分布最中性,从而减弱了与生物污染物的静电/偶极相互作用。
相比之下,CBAs因间隔基增长,偶极矩和静电势显著增强,不利于抗污染性能。此外,静电势与部分电荷分析表明,TMAO的氧原子部分电荷(−0.48)远低于CBAs(−0.91),促进了更强的氢键(部分共价性)和水合作用。
这些结果为理解两性离子的水合稳定性及抗污染性能的分子设计提供了关键电学依据。
2. HOMO/LUMO能级
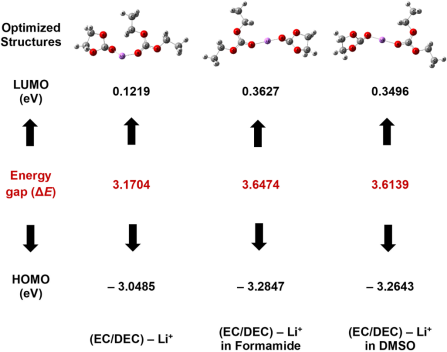
通过计算HOMO-LUMO,揭示了EC/DEC基电解质在不同溶剂化环境(甲酰胺、DMSO)中的电化学稳定性差异。
通过对比纯EC/DEC(ΔE=8.3604 eV)与含添加剂体系(甲酰胺:8.6663 eV;DMSO:8.6680 eV)的能隙,研究者发现添加剂通过增大HOMO-LUMO能隙显著提升了电解质的氧化还原稳定性。
HOMO能级关联阴极反应倾向,LUMO能级关联阳极反应倾向,能隙扩大表明添加剂削弱了溶剂分子与阳离子的轨道相互作用,使其更难发生氧化/还原反应。
该计算从电子结构层面阐明了溶剂化结构对电解质稳定性的调控机制,为设计高稳定性电解液提供了理论依据。
3. 键长与键角
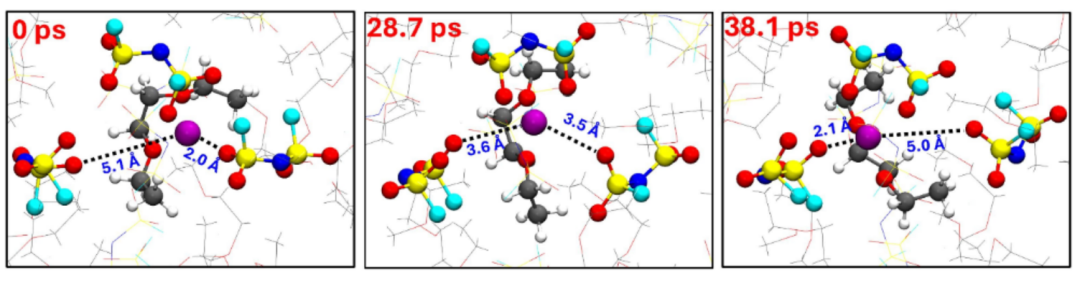
在下图中, Li⁺与FSI⁻的配位键长变化(0-38.1 ps)直接关联离子解离动力学:当左侧FSI⁻接近Li⁺(28.7 ps)时引发配体交换,右侧FSI⁻随即解离(键长增大至38.1 ps),而Li⁺始终与DEE保持配位(稳定键长)。
这种键长演变证实弱溶剂化电解液能促进Li⁺快速脱离阴离子(FSI⁻解离能垒低),但溶剂(DEE)配位键的持续性导致不完全解离。
该发现为设计“弱溶剂化–快速解离“电解液提供了结构动力学依据,通过优化键长稳定性可同步提升Li⁺迁移率和界面脱溶剂效率,对高功率电池开发具有指导意义。
4. 溶剂化能与去溶剂化能
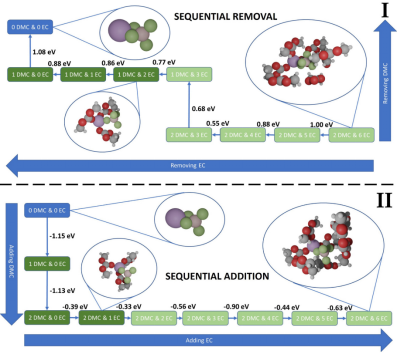
在下图中,溶剂化能计算被用于揭示LiBF₄在EC/DMC混合电解液中的溶剂化结构动态演变规律及其热力学稳定性。
通过从头算分子动力学(AIMD)结合簇模型分),研究发现Li⁺优先与DMC的羰基氧结合(-1.15 eV vs EC的-1.06 eV),但受DMC甲基空间位阻影响,最终溶剂化壳层形成2个DMC和6个EC的配位结构。
溶剂化能计算量化了分子移除/添加过程的能量变化(0.33-1.15 eV),揭示BF₄⁻侧溶剂分子更易解离(0.55 eV),而Li⁺侧因强静电作用(Li-O电负性差异)需更高能量(达1.08 eV)。
该研究通过溶剂化能与空间位阻的协同分析,阐明了动力学模拟对真实溶剂化结构重构的关键作用,为优化锂盐溶剂化环境提供了能量学依据。
5. 结合能(Binding Energy):
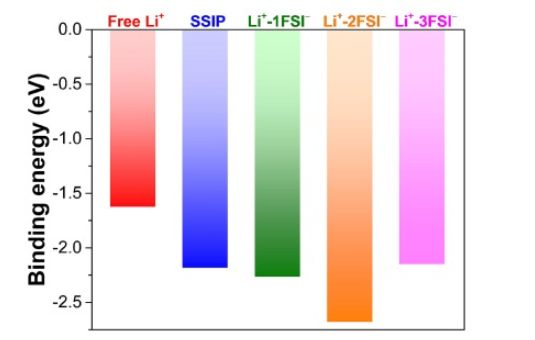
在下图中,结合能计算被用于揭示LiFSI-DEE电解液在不同浓度下的锂离子溶剂化结构稳定性差异。
通过DFT计算结合隐式溶剂模型(C-PCM),研究发现低浓度下溶剂分离离子对(SSIP)结构因FSI⁻位于次级溶剂化壳层而表现出显著稳定性(结合能比自由Li⁺更低),这与RDF中Li-FSI次级峰增强的现象相印证;而高浓度下Li⁺-2FSI⁻聚集体(AGG-2)因电荷分布平衡成为最稳定构型。
静电势(ESP)分析进一步显示,DEE的给电子特性导致自由Li⁺结构存在静电排斥,而AGG-3因阴离子间排斥作用稳定性下降。
该研究通过结合能与ESP的关联分析,首次量化了次级溶剂化壳层分子对锂离子配位结构稳定性的关键影响,为调控电解液浓度依赖的溶剂化行为提供了理论依据。
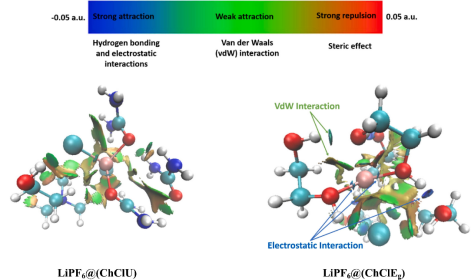
6. 相互作用
通过非共价相互作用(NCI)分析结合电子密度梯度(RDG)方法,系统研究了LiPF₆和LiTf₂N在深共晶溶剂(DES)中的溶剂化结构及其相互作用机制。
研究显示,锂离子与溶剂分子间的C-O···Li⁺、HO···Li⁺等相互作用呈现亮蓝色等值面,证实了这些作用以强静电主导,与前期原子–原子相互作用(ADI)分析结果一致。
同时,氢键给体(HBDs)和胆碱阳离子(Ch⁺)通过蓝色(静电)至绿色(范德华力)的等值面与第一溶剂化层(FSS)形成多尺度相互作用网络。
该分析不仅明确了Li⁺溶剂化壳层的静电作用优势,还揭示了弱相互作用(如CF₃(S-O)₂···Li⁺的范德华力)对复合物稳定的协同贡献,为优化电解质溶剂化结构设计提供了原子尺度的相互作用图谱。