

水分子研究在《Nature》和《Science》等顶级期刊上的高频出现绝非偶然,其背后蕴含的深层次科学逻辑与跨学科价值构成了这一现象的核心驱动力。从分子层面的独特物化性质到宏观尺度的生态系统功能,水的研究始终处于科学前沿的交叉点,不断催生突破性发现。
以下从多维度展开分析,结合具体实验图像与机制图解,揭示其持续占据学术顶刊的必然性。
水分子的基础物化特性
水分子(H₂O)的简单结构掩盖了其物理化学性质的非凡复杂性。水分子的弯曲构型(键角≈105°)和极性共价键导致氧原子带部分负电荷(δ⁻),氢原子带部分正电荷(δ⁺),形成强偶极矩(1.8 D)。这种极性使其成为自然界最优的氢键形成者:每个水分子最多可与4个相邻分子形成动态氢键网络。
在纳米通道内,水分子排列成准一维链状结构,氢键的协同振动使质子以超快速度传递(>10⁹ s⁻¹),成为生物水通道蛋白(如AQP1)高效输水的结构基础。2003年诺贝尔化学奖授予Peter Agre的发现正是基于此,揭示了生命体内水分子的定向传输机制。
水氢键网络的动态重组还直接解释了其40余项物性异常
密度异常:冰的密度低于液态水(4℃时密度最大),源于固态下氢键延展形成的六方晶格。冰浮于水面形成隔热层,保护水生生态系统越冬生存。
热容与相变潜热异常:水的比热容(4.18 J/g·K)和汽化热(2260 kJ/kg)远高于多数溶剂,因其氢键断裂需额外能量。水分子在气液界面形成致密氢键层,产生72.8 mN/m的高表面张力(仅次于汞),使水滴呈球状,昆虫得以在水面行走。
溶剂普适性:极性水分子的强溶剂化能力使其成为“万能溶剂”,溶解离子化合物(如NaCl)时,水分子通过偶极作用拆解晶格,形成水合离子壳层,驱动生物体内的营养输送与代谢。
案例分享
The effect of hydration number on the interfacial transport of sodium ions
研究团队借助超高分辨率扫描隧道显微镜(STM)与qPlus非接触原子力显微镜(AFM)的组合系统,在NaCl (001) 表面通过逐步添加单个氘水分子(D₂O)的方式,构建了水合数n=1 至 5 的钠离子水合物(Na⁺・nD₂O)。
实验中利用Cl⁻修饰的探针操控离子水合物,并通过非弹性电子隧穿技术在低温(5 K)下激活其扩散,结合STM/AFM图像与分子力学模型模拟,精准表征了不同水合数离子的吸附位点、结构特征及扩散行为。研究发现,三水合钠离子(Na⁺・3D₂O)的扩散速率比其他水合物快数个数量级。
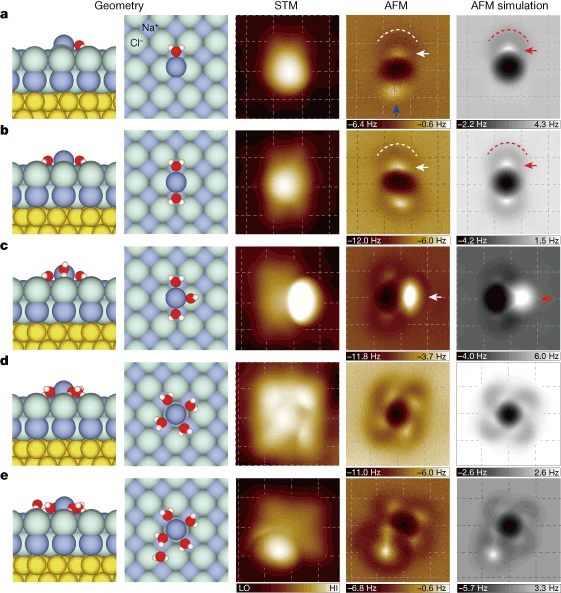

DOI:10.1038/s41586-018-0122-2
密度泛函理论(DFT)计算揭示了三水合钠离子扩散势垒极低()的根源——其存在亚稳态,三个水分子可集体旋转且能垒极小,而其他水合物的扩散势垒均高于200 meV。
经典分子动力学模拟进一步证实,该水合物在室温下仍具有超高迁移率,其扩散行为与水合物和表面晶格的对称性匹配程度密切相关。理论计算从原子尺度阐释了水合数对离子迁移率的调控机制,为通过界面对称性工程优化纳米流体系统中的离子传输提供了理论依据。
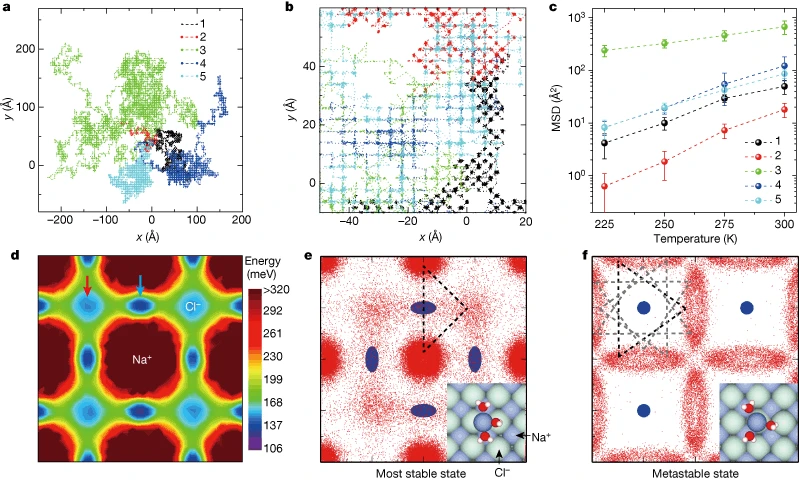
DOI:10.1038/s41586-018-0122-2
In situ Raman spectroscopy reveals the structure and dissociation of interfacial water
研究团队运用原位拉曼光谱(SHINERS)、电化学技术以及从头算分子动力学(AIMD)模拟,对原子级平整的Pd单晶表面的界面水结构与解离过程展开研究。
借助三电极体系和超薄溶液层设计,结合壳层隔绝纳米粒子增强拉曼技术,在0.1 M NaClO₄溶液中对不同电位下的界面水进行原位探测,发现界面水由氢键水和Na⁺水合物构成,且在析氢反应(HER)电位下,受偏压和Na⁺协同作用,界面水从无序分布转变为有序结构,进而提升HER速率。

DOI:10.1038/s41586-021-04068-z
AIMD 模拟从原子尺度阐释了界面水结构有序化的机制,证实Na⁺水合物(Na・H₂O)在负电位下会使Pd-H距离缩短,促进费米能级与水的反键轨道相互作用,从而增强电子转移效率,加速水的解离。
此外,模拟还揭示了界面水在电场作用下从无序到有序的动态重组过程,以及Na⁺浓度对水结构有序性和HER活性的影响,为通过局部阳离子调控优化界面水结构以提升电催化反应速率提供了理论支撑。
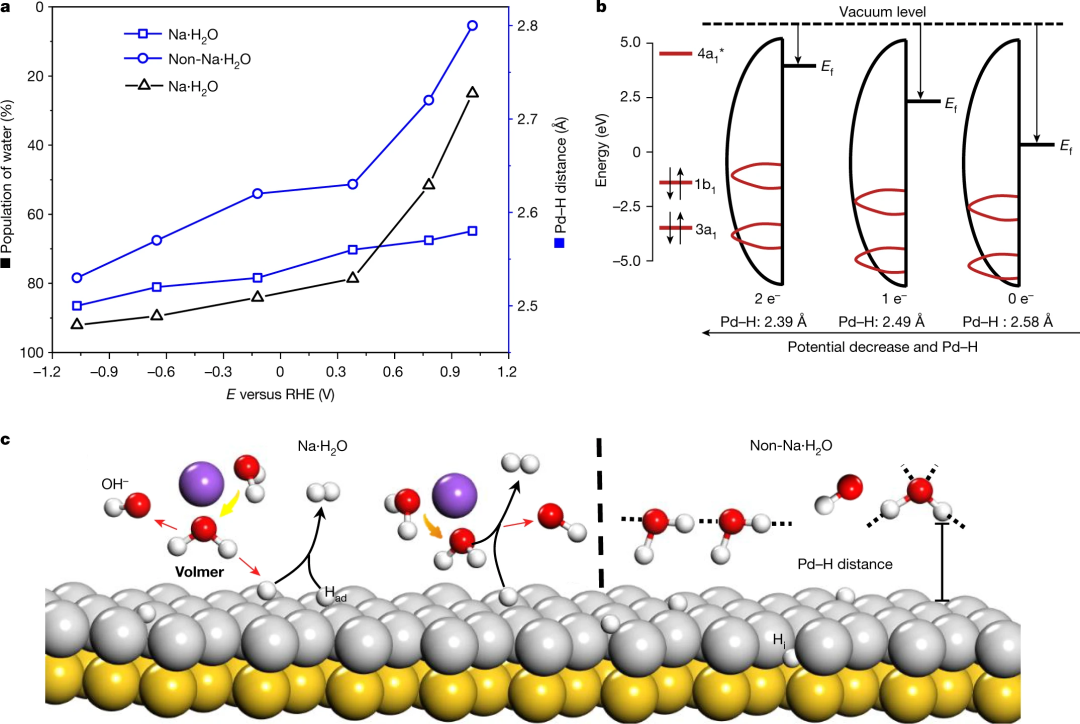
DOI:10.1038/s41586-021-04068-z
Imaging the short-lived hydroxyl-hydronium pair in ionized liquid water
研究团队运用液相超快电子衍射技术(MeV-UED),以800nm激光脉冲电离约100nm厚的液态水层,通过测量动量空间0.5至11.5 Å⁻¹的衍射信号并转换为原子对分布函数(PDF),实时追踪电离水的分子结构动态。
实验捕捉到电离后140飞秒内形成的短寿命羟基–水合氢离子自由基阳离子对(OH (H₃O⁺)),其通过氧・・・氧键收缩和质子转移形成,并在250飞秒内解离,随后水结构弛豫。该技术直接探测到 O・・・O和O・・・H键的变化,为电离水初始反应路径提供了直接结构证据。
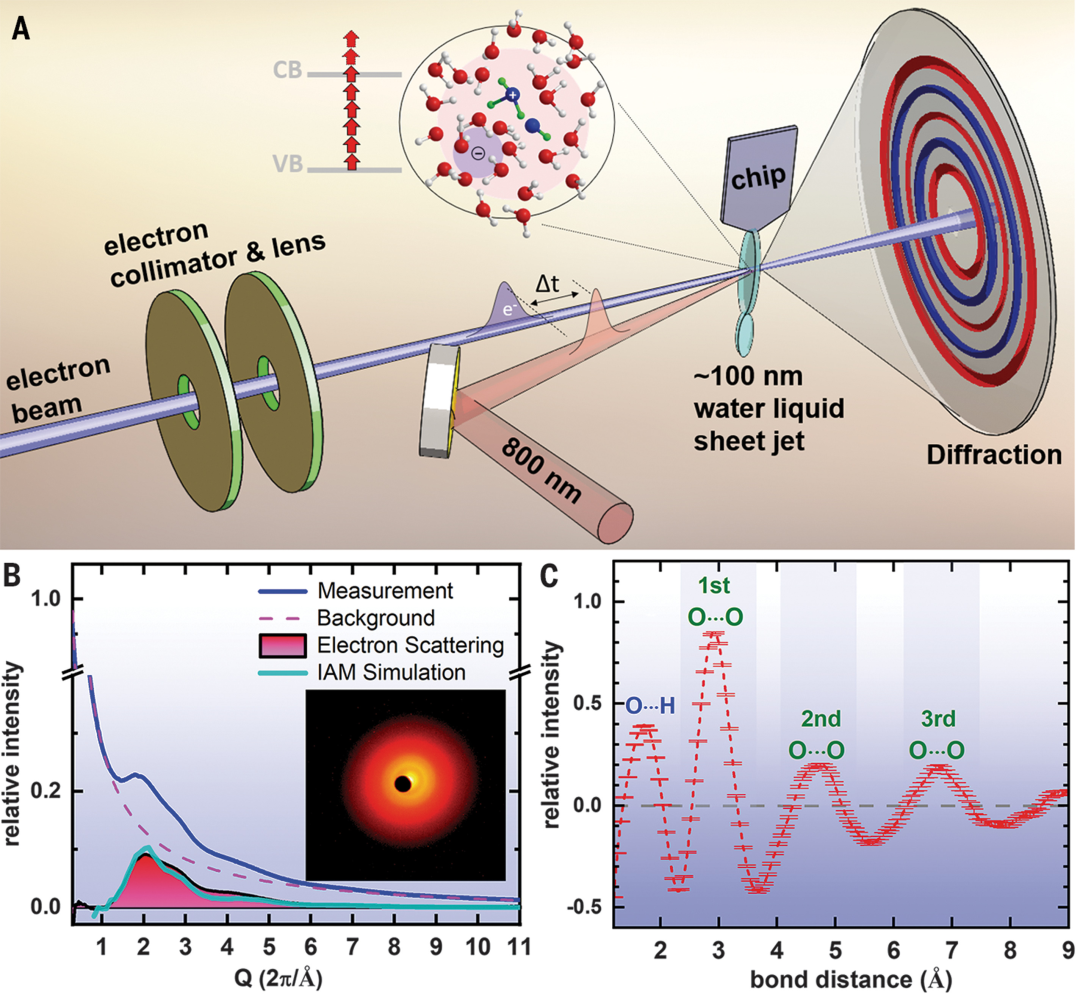
DOI: 10.1126/science.abg3091
从头算分子动力学(ab initio MD)模拟验证了OH (H₃O⁺) 对的瞬态结构,其预测的O・・・O和O・・・H键距离与实验结果一致,揭示了质子转移过程中键长收缩的机制。时间依赖密度泛函理论(TD-DFT)模拟估算了激光脉冲在水中的能量沉积,预测局部温度升高约320K,解释了电离后水结构弛豫类似热加热的现象。
此外,分子动力学(MD)模拟通过独立原子模型(IAM)拟合电子衍射数据,辅助解析了液态水在电离后的结构动态响应,为理解电离水的非辐射弛豫和热效应提供了理论支撑。

DOI: 10.1126/science.abg3091
总结
水分子持续占据《Nature》《Science》的核心位置,本质源于其三重属性。
基础性:氢键网络的独特物化性质是理解生命、材料及自然现象的物理基础;
交叉性:从纳米生物到行星科学,水连接多学科前沿问题;
工具依赖性:冷冻电镜、超快光谱等技术突破始终以水为试金石。
据统计,近十年水相关顶刊论文年均增长17.3%,2025年仅恒星水冰研究即引发15篇Nature子刊评论。未来,随着量子传感、原位电镜等技术发展,水分子的量子效应与界面行为将继续成为顶刊的“常驻主题”,推动人类对物质世界认知的边界。