


同步辐射XAFS实验样品的厚度/重量如何确定?选透射还是荧光?SAMPLEM4M一键解决!!
精选干货|同步辐射PDF基础知识及经典应用分析!
同步辐射XAFS技术对催化剂的微观世界有着敏锐的洞察力。它对吸收原子周围的局域结构和化学环境极为敏感,能够精准地描绘出吸收原子周围的配位结构,以及原子的价态和电子结构等关键信息。
这种技术的一大亮点在于,它对样品的形态没有过多要求,无论是聚集态还是结晶态,无论是固态还是液态,都能进行测试。
更重要的是,它可以在不破坏样品的情况下,进行原位测量,这对于研究真实反应状态下的催化材料体系的结构演变来说,简直是“量身定制”的利器。
原位XAFS技术就像是一个高精度的“显微镜”,能够深入催化剂的原子和电子结构内部。
通过这种技术,可以实时观察催化剂在反应过程中的动态变化,从而更好地理解催化剂的结构与性能之间的内在联系。这种深入理解,不仅能优化现有催化剂的性能,还能为设计和制备新型催化剂提供重要的理论依据。

中科大姚涛教授团队通过掺杂高价铬制备了一系列具有高性能AEMWEs的非晶态金属氧化物催化剂(FeCrOx、CoCrOx和NiCrOx)。
通过同步辐射原位技术发现,Co位点从低价态到高价态的转变对中间体吸附能和较低的氧化势垒的积极影响是其活性得到提高的关键因素。
为比较和探讨CoCrOx具有优异活性的关键原因,通过X射线近边结构吸收光谱(XANES),确定了活性元素的价态。
将吸收边的位置与标准样品进行比较,发现CoCrOx位于CoO和Co3O4之间(图a)。所有催化剂的Cr k边几乎与Cr2O3重叠,表明Cr的价态为+3(图b),这与sXAS和XPS表征结果一致。
为进一步阐明三种催化剂内在催化活性差异的原因,使用sXAS表征FeCrOx、CoCrOx和NiCrOx在反应前后活性元素的价态变化(图c)。
由于Co2+的结合能高于Co3+,研究发现,Co吸收峰向高能量方向移动1.9 eV,表明Co的氧化态明显增加。Co2+的特征卫星峰几乎消失,进一步证明Co2+在OER过程中几乎被完全氧化到较高的价态。
为揭示电催化OER过程中Co活性位点的电子结构和原子局部环境的动态演变,进行原位XAFS和vtc-XES表征。如图d所示,Co的K边XANES谱表明Co在非原位和开路电压(OCV)条件下的吸收边位置几乎相同。
施加电压时,Co的近边吸收边正移位,表明在OER过程中Co的平均氧化态升高,这与sXAS和XPS结果一致。
Co K边 EXAFS谱的拟合结果表明,随着电压的施加,第一壳层(Co-O)的配位数从4显著增加到6以上(图e),配位数的增加可能是Co价态升高的原因。图f显示了吸收边位置与Co-O配位数之间存在显著的正相关关系。
此外,Co的局部结构在反应过程中发生了变化,这可能是CoCrOx的动态重构。
DOI:10.1038/s41467-024-47736-0
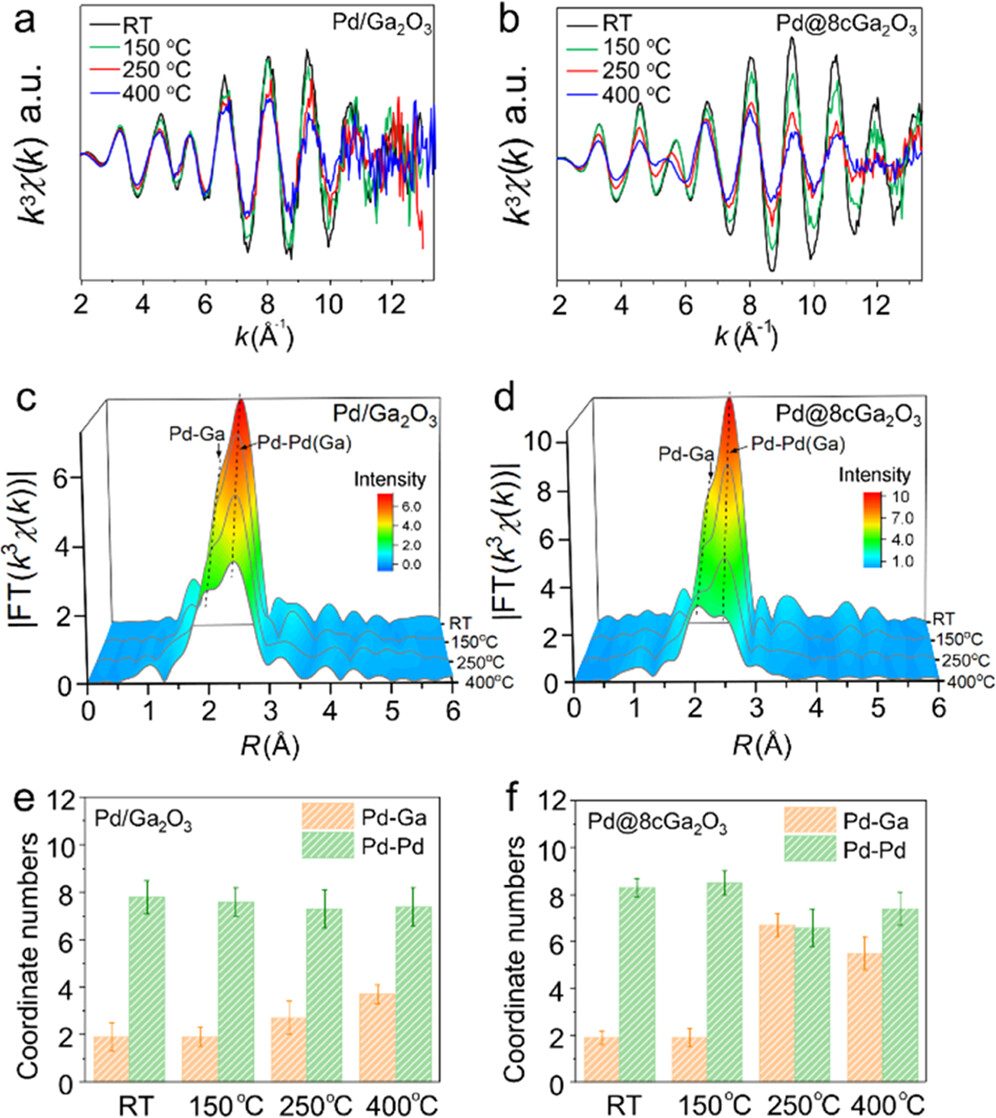
CO2加氢反应中,高效催化剂的设计是重点,因此利用XAFS技术认识催化反应的真实活性位点以及建立和理解催化反应构效关系至关重要。
中国科学技术大学路军岭团队,利用原位XAFS技术,探究了在CO2加氢反应条件下,不同负载量的Pd/Ga2O3催化剂结构随着还原温度的升高而发生变化。
随着还原温度的升高, 5%Pd@8cGa2O3与5%Pd@8cGa2O3催化剂中的Pd均表现相似的金属态。
与5%Pd/Ga2O3相比,5%Pd@8cGa2O3的振荡幅度比5%Pd/Ga2O3的振荡幅度下降得更快(图a, b), 这意味着5%Pd@8cGa2O3中Pd元素的配位下降更急剧,EXAFS光谱的傅里叶变换(FTs)也证实了这点(图c, d),具体表现为Pd–Pd配位数明显减少,而Pd–Ga配位数迅速增加。
对于5% Pd@8cGa2O3,他们观察到在250℃下经过H2还原后,Pd–Ga配位数从1.9迅速增加到6.7,同时Pd–Pd配位数从8.3大幅减少到6.2,证实了富Ga合金相的形成;随着还原温度升高至400℃,Pd–Pd与Pd–Ga配位数与标样Pd2Ga中对应的配位数接近,表明形成了Pd2Ga合金相。
这些发现为将RMSI扩展到低温加氢反应领域提供了新的途径, 使得制造用于高效催化的新金属合金相成为可能。
DOI:10.1021/jacs.2c12046

作为多用途化学中间体的苯甲醛,在制药工程、香水制备和精细化工生产等领域有着广泛的应用,因此采用苯甲醇催化氧化法制备高纯度苯甲醛在工业中受到重视。通过催化生产高纯度苯甲醛的关键在于开发性能优异的催化剂。
苏黎世联邦理工学院Alfons Baiker教授将原位XAFS和衰减全反射红外光谱(ATR-IR)相结合,研究了Bi对0.75 wt% Bi-5 wt% Pd/Al2O3催化剂液相好氧氧化过程中表面物质演化和结构的影响。
XAFS结果表明,在该条件下,Bi和Pd都处于还原态。在有氧条件下,ATR-IR和XAFS都表明Bi控制了贵金属的氧气供应。此外, 在不同实验条件下,Pd都保持金属态。
这是因为Bi使得Pd/Al2O3催化剂具有更强的抗过氧化能力,因此在存在过量氧气的情况下,其活性时间更长。
最后, 在Bi的存在下,苯甲醛水合/氧化生成羧酸盐受到很大阻碍。这项研究展示了两种技术结合的潜力,通过XAFS得出粒子结构/氧化态,通过ATR-IR确定表面物质的结论,从而实现结构与催化性能相关联。
DOI:10.1016/j.jcat.2007.09.013
析氧反应(OER)涉及多电子转移过程,动力学反应较慢,揭示其催化反应机理对于提高电催化剂活性、稳定性和降低能源转换成本至关重要。
中国科学技术大学韦世强团队借助原位XAFS实验技术从原子级别表征了Ir单原子催化剂在电催化析氧反应过程中的真实活性结构及其动态演变。

他们通过电驱动氨基沉积策略开发了异氮配位的Ir单原子催化剂(AD-HN-Ir)并用于催化酸性OER。
原位XANES表征结果如图所示,在反应电位驱动下电子从Ir位点向周围配位原子转移,金属Ir的电子空位增加。
图中的EXAFS图谱显示,在预催化阶段的1.25 V电位下,非金属配位壳层(Ir-N/O)的峰值强度与非原位状态相比有所增强。
相应的XAFS拟合分析显示,除了初始的Ir-N4构型外,Ir位点上出现一个与氧吸附有关的Ir–O键(图c)。
当电位升高到1.35和1.45 V时,Ir–N/O配位峰的强度进一步增强,扩展边中相应的峰位置出现偏移,这可能与吸附在单位点上的羟基进一步演化形成的*OOH中间体有关。该原位表征深入解析了O-hetero-Ir-N4单原子活性位点在催化OER的动态作用过程。
DOI: 10.1038/s41467-021-26416-3